Il existe plus de 200 cardiopathies congénitales allant de la plus simple à la plus complexe. Beaucoup d’entre elles passent inaperçues lors des échographies de grossesse. Il n’est donc pas ici question de tout voir mais de dépister les plus graves ayant une incidence sur la prise en charge.
I-La circulation du sang pendant la grossesse et les changements à la naissance (adaptation néonatale)
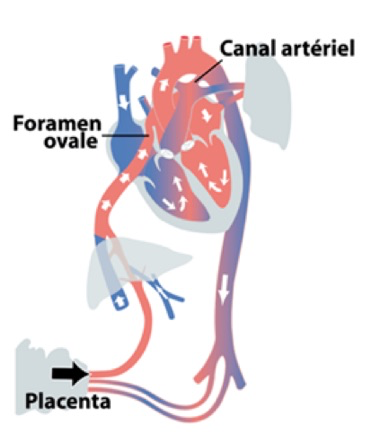
Avant la naissance, les poumons du fœtus ne sont pas fonctionnels et n’interviennent pas dans l’oxygénation du sang. C’est le placenta qui remplit ce devoir d’oxygénation. Le sang oxygéné du placenta se mélange au sang bleu du fœtus au niveau de la veine cave inférieure, avant d’atteindre l’oreillette droite. Etant donné que les poumons du fœtus sont non fonctionnels, peu de sang y est déversé. Grâce au foramen ovale et au canal artériel, 2 structures spécifiques du foetus, le cœur droit (avec son sang oxygéné en provenance du placenta) peut se déverser directement dans la circulation gauche en court-circuitant ainsi les poumons. En effet, le foramen ovale est une large fente, située dans le septum interauriculaire. Il permet le passage du sang de l’oreillette droite, surtout celui en provenance de la veine cave inférieure (et donc du placenta) vers l’oreillette gauche. Le canal artériel est un « vaisseau provisoire » très bref qui relie l’artère pulmonaire à l’aorte. Ces 2 structures permettent ainsi au sang oxygéné de passer vers l’aorte et ainsi vers le cerveau mais également les autres organes. On voit ainsi que grâce à ces 2 structures et sans aucun bouleversement anatomique, le fœtus assume une fonction circulatoire complètement différente de celle du nouveau-né. Cette situation explique pourquoi la plupart des malformations cardiaques sont fort bien tolérées in utero : la circulation pulmonaire n’est pas existante et l’organisation ventriculaire n’a pas d’importance puisqu’un seul ventricule (le droit en situation normale) suffit à assumer la circulation du circuit ‘unique’.
A la naissance le placenta se sépare brusquement de la circulation et l’enfant doit trouver d’urgence une autre source d’oxygène : il lui faut utiliser ses poumons ! Grâce à ces premiers mouvements respiratoires, les poumons et les vaisseaux pulmonaires s’ouvrent. Cette ouverture des vaisseaux engendre une baisse des ‘résistances» pulmonaires, autrement dit, le sang y passe beaucoup plus facilement, avec moins de «résistance’. Ceci permet au sang de perfuser les poumons et de s’oxygéner. Le sang oxygéné arrive ensuite en grande quantité des poumons dans l’oreillette gauche, ce qui y augmente la pression et ferme la fente du foramen ovale : la circulation pulmonaire est installée et est définitivement séparée de la circulation systémique lorsque le canal artériel se ferme quelques heures plus tard suite au passage d’un sang plus oxygéné. A noter que la baisse des «résistances» pulmonaires, phénomène très important dans l’adaptation néonatale, se fait en grande partie à la naissance mais se poursuit encore pendant quelques semaines.
II-Les anomalies visibles sur la coupe des 4 cavités
A-Ventricule unique
Le terme «cœur univentriculaire» ou «ventricule unique» regroupe un grand nombre de malformations cardiaques différentes qui ont en commun le fait que un des 2 ventricules ne s’est pas ou insuffisamment développé et ne pourra donc pas être utilisé pour le fonctionnement du cœur. En présence d’un seul ventricule, le sang rouge et bleu se mélange obligatoirement et le travail de ce ventricule est augmenté.
La fréquence des malformations de type ventricule unique est difficile à déterminer car ce groupe reprend un grand nombre de malformations différentes. On peut estimer cependant qu’il représente moins de 10% de l’ensemble des malformations cardiaques congénitales
Diverses classifications existent. En fonction de la présentation clinique initiale, on peut distinguer les ventricules uniques avec ou sans rétrécissement pulmonaire, c’est-à-dire obstacle sur la voie pulmonaire (au niveau, en dessous et/ou au dessus de la valve) gênant le passage du sang entre le ventricule et les artères pulmonaires. La présence d’un rétrécissement pulmonaire, engendre une cyanose plus sévère alors qu’en absence de rétrécissement pulmonaire le tableau clinique est celui d’une défaillance cardiaque par excès de perfusion pulmonaire.
Les ventricules uniques peuvent également être distingués en fonction du type anatomique. De ce point de vue là, les 3 types les plus fréquemment rencontrés sont: l’atrésie tricuspide, le ventricule gauche à double entrée et l’hypoplasie du cœur gauche.
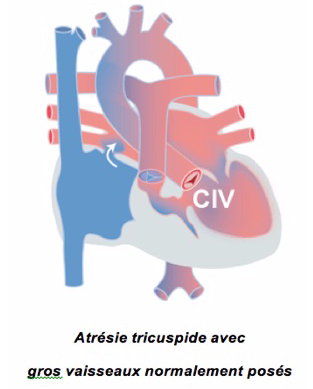
1-L’atrésie tricuspide
La valve tricuspide et le ventricule droit ne se sont pas développés. Le sang bleu doit donc obligatoirement passer de l’oreillette droite dans l’oreillette gauche où il se mélange au sang rouge venant des poumons. Ce sang mélangé passe dans le ventricule gauche qui éjecte aussi bien dans l’aorte que dans l’artère pulmonaire, via une communication interventriculaire (CIV) et/ou par le canal artériel resté ouvert (figure 1). Dans cette malformation, les gros vaisseaux peuvent être normalement posés dans quel cas il y a souvent une sténose pulmonaire, limitant le passage de sang du ventricule vers les artères pulmonaires. Les vaisseaux peuvent par contre être ‘transposés’, c’est à dire que l’aorte naît du petit ventricule droit et l’artère pulmonaire naît du ventricule gauche. Dans ces cas, l’aorte est souvent plus petite et une coarctation de l’aorte est souvent présente.
2-Le ventricule gauche à double entrée
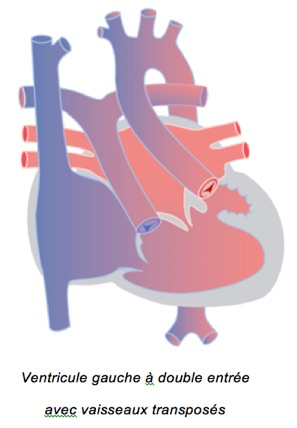
Les valves tricuspide et mitrale sont toutes les deux orientées vers le ventricule unique gauche. Le petit ventricule ‘droit’, souvent situé à gauche (NB un ventricule est définit comme gauche ou droit en fonction de sa morphologie et non en fonction de sa position !), ne participe quasi pas à la circulation. La transposition des grands vaisseaux est souvent associée (mais pas toujours): l’aorte naît du petit ventricule droit, l’artère pulmonaire naît du grand ventricule gauche. D’autres malformations peuvent être présentes comme une sténose pulmonaire, une coarctation, des anomalies des veines pulmonaires, etc.
3-L’hypoplasie du cœur gauche
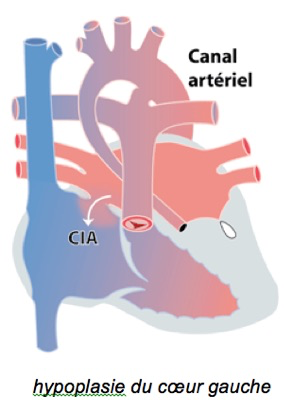
Dans ce cas, toute la partie ‘gauche’ du cœur ne s’est pas ou mal développée: la valve mitrale, le ventricule gauche, l’aorte (figure 3). Le ventricule droit doit faire l’entièreté du travail: éjecter le sang vers l’artère pulmonaire mais également vers l’aorte, et ce grâce au canal artériel (CA). Ici aussi, il peut y avoir d’autres lésions associées comme une coarctation, une anomalie du retour veineux pulmonaire etc..
Beaucoup d’autres formes anatomiques existent, mais il est impossible de toutes les décrire. La présentation clinique, la prise en charge et le pronostic varient d’une malformation à l’autre, mais il existe néanmoins des grandes ‘similitudes’ entre ces différentes malformations. Ceci nous permet donc de vous donner une description assez ‘générale’ de comment un enfant se présente, comment il est pris en charge et ce que l’on peut espérer pour son avenir.
L’avantage du dépistage in utero est de permettre l’organisation de la naissance de l’enfant dans un centre où il peut être pris en charge immédiatement (parfois en salle d’accouchement même) par une équipe de cardiologie pédiatrique et ce, sans devoir transférer l’enfant malade (et la maman) d’un hôpital à l’autre.
B-Dilatation auriculaire droite
Devant une dilatation de l’oreillette deroite, il faut rechercher une anomalie de la valve tricuspide. Sil elle est normale il s’agit d’une exceptionnelle dilatation idiopathique de l’oreillette. Dans le cas contraire, s’il existe une fuite, on évoque la maladie d’Ebstein ou une dysplasie valvulaire tricuspide. Ces cardiopathies peuvent s’accompagner d’une cardiomégalie massive d’une anasarque et de troubles du rythme.
1-La maladie d’Ebstein ou malformation d’Ebstein
C’est une cardiopathie congénitale rare (1% des cardiopathies) caractérisée par le déplacement des feuillets septal et inférieur de la valve tricuspide, normalement situés au niveau de la jonction auriculo-ventriculaire, vers la pointe du ventricule droit.
Normalement, la valve tricuspide est constituée de trois feuillets (septal, antérieur et postérieur), insérés sur l’anneau auriculo-ventriculaire et s’ouvrant sensiblement dans le même plan que la valve mitrale, son homologue située entre l’oreillette gauche et le ventricule gauche.
Dans la malformation d’Ebstein, l’ouverture des feuillets septal et postérieur est déplacée vers la pointe du ventricule droit, à une distance plus ou moins grande de l’anneau auriculo-ventriculaire et il s’associe habituellement un certain degré de malformation du 3° feuillet antérieur. Cette anomalie résulte d’un défaut de clivage de la valve au cours de la vie fœtale. Pour plus de détail, consulter ci-dessous le paragraphe consacré à l’embryologie de cette malformation.
En plus de cette anomalie d’implantation, la valve tricuspide malformée est le siège d’une fuite quasiment constante mais d’intensité variable. Dans 1/4 des cas, il s’y associe un rétrécissement valvulaire.
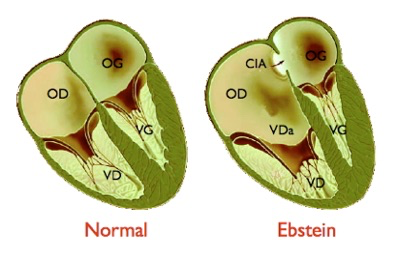
Cette anomalie est responsable d’une modification de l’anatomie fonctionnelle du cœur droit. Normalement, l’oreillette droite (OD) et le ventricule droit (VD) se contractent successivement, la contraction de l’oreillette se produisant alors que la valve tricuspide est ouverte, celle du ventricule droit après qu’elle s’est fermée, pour éviter un reflux de sang dans l’oreillette. L’implantation trop basse de la valve tricuspide modifie la répartition des cavités. L’oreillette droite est dilatée, formée de l’oreillette normale et de la partie proximale du ventricule droit (portion « auricularisée » – VDa sur le schéma). Cette oreillette a une cinétique anormale, avec une double contraction se produisant aux temps auriculaire et ventriculaire du cycle cardiaque. Le ventricule droit est quant à lui de dimensions réduites, au prorata de l’importance du déplacement de la valve tricuspide.
Donc, maladie d’Ebstein =
Dilatation tricuspide + fuite tricuspide
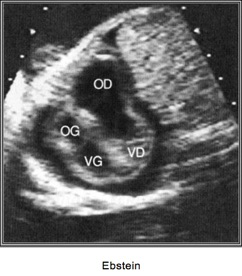
– Déplacement du feuillet septal de la valve tricuspide vers l’apex du VD
– Souvent associés à des anomalies morphologiques des feuillets valvulaires et à une insuffisance tricuspide de sévérité variable.
– Dilatation OD + parfois dilatation VD (due à la fuite valvulaire)
Dans un tiers à la moitié des cas, d’autres malformations cardiaques lui sont associées:
• Les plus fréquentes sont :
-CIA sur le schéma ou d’un foramen ovale perméable observée dans 50 % des cas au moins, (90 % pour certains auteurs) et qui modifie profondément la symptomatologie en transformant l’anomalie d’Ebstein en cardiopathie cyanogène par passage anormal de sang de l’oreillette droite dans l’oreillette gauche.
-La persistance de voies de conduction électrophysiologiques anormales entre les oreillettes et les ventricules, qualifiées également de « voies accessoires », susceptibles de se traduire par un Syndrome de Wolf-parkinson-White sur l’ECG et de provoquer des troubles du rythme cardiaque (25 % des cas au moins).
-Une sténose pulmonaire qui aggrave considérablement le pronostic.
Dans les associations plus rares, signalons celle d’une anomalie d’Ebstein et d’une transposition corrigée des gros vaisseaux, deux malformations pourtant exceptionnelles.
Remarques:
• L’anomalie d’Ebstein est loin d’être univoque. Lorsque le défaut de clivage est minime, elle est souvent méconnue car sans aucun retentissement, le cœur fonctionnant dans des conditions quasi normales. À l’opposé, dans les formes majeures, la cavité ventriculaire droite est extrêmement réduite et incapable d’assurer la fonction qui lui est normalement dévolue. Entre ces deux extrêmes, tous les intermédiaires sont possibles.
• L’anomalie d’Ebstein n’est pas associée à une anomalie chromosomique spécifique. Quelques formes familiales ont été décrites. Il n’y a pas de conseil génétique spécifique à cette cardiopathie.
2- Dans la dysplasie tricuspide,
on a :Dilatation tricuspide + fuite tricuspide. Les feuillets valvulaires anormaux, redondants, épaissis mais l’insertion du feuillet septal de la tricuspide est normale. Dilatation OD + parfois dilatation VD (due à la fuite valvulaire)
C-Solution de continuité dans les cloisons inter-auriculaire et inter-ventriculaire
1- Communications inter-auriculaires
Diagnostic anténatal difficile car existence obligatoire d’un shunt inter-auriculaire. Le septum primum correspond à la valvule de Vieussens qui est facilement mise en évidence dans l’OG. La cloison inter-auriculaire est perforée en son centre par le foramen ovale.
Une CIA type ostium secondum peut être suspectée lorsque la valvule de Vieussens est absente ou trop peu développée pour recouvrir en totalité la surface du foramen ovale.
La CIA de type ostium primum est assimilée aux canaux atrio-ventriculaires partiels et est caractérisée par l’absence de la partie basse du septum inter-auriculaire située au contact des valves auriculo-ventriculaires. Ces dernières sont situées à la même hauteur et l’insertion physiologique un peu plus apicale du feuillet septal de la tricuspide par rapport à son homologue contro-latéral n’est pas retrouvée. Il s’y associe habituellement une insuffisance mitrale liée à une fente de la grande valve mitrale.
2-Communications inter-ventriculaires
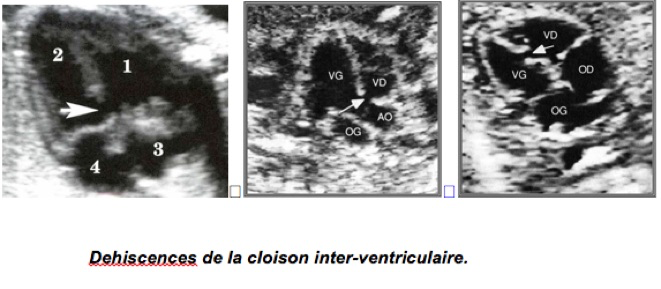
Isolées dans 60 % des cas.
Echocardiographie bidimensionnelle :
Interruption localisée du septum inter-ventriculaire.
L’incidence des quatre cavités est souvent prise à défaut pour le diagnostic des CIV située sous les gros vaisseaux (les plus fréquentes).
Celles-ci ne sont le plus souvent visibles que dans les incidences longitudinales par l’aorte ou l’AP.
Confirmation de la CIV par le doppler couleur.
Le doppler couleur permet de visualiser de petites CIV musculaires situées au centre du septum inter-ventriculaire ou à l’apex du cœur, non visibles en échographie bidimensionnelle.
Les CIV larges doivent faire rechercher :
-une malformation associée cardiaque ou extra-cardiaque
-une anomalie chromosomique
Les petite CIV musculaire isolée peuvent se refermer spontanément avant la naissance dans 50 % des cas.
D-Canal atrio-ventriculaire
Ensemble de malformations concernant un défaut de développement du septum inter-auriculaire, du septum inter-ventriculaire et des valves auriculo-ventriculaires. On distingue:
1-Canal atrio-ventriculaire complet :
– Large défaut septal concernant à la fois les cloisons inter-auriculaire et inter-ventriculaire
– Valve auriculo-ventriculaire commune qui connecte les deux oreillettes aux ventricules
En diastole, la valve auriculo-ventriculaire commune s’ouvre dans les deux ventricules.
En systole, la croix du cœur n’existe pas, de part la présence d’une CIV du septum d’admission et d’une CIA située au contact de la valve AV commune
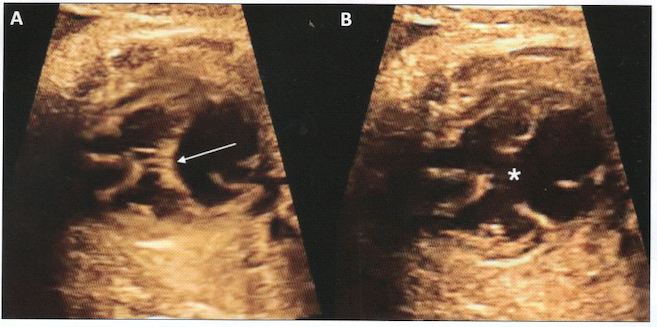
A:systole; valve unique fermée (flèche)
B-diastole: défectueuse central majeur (*)
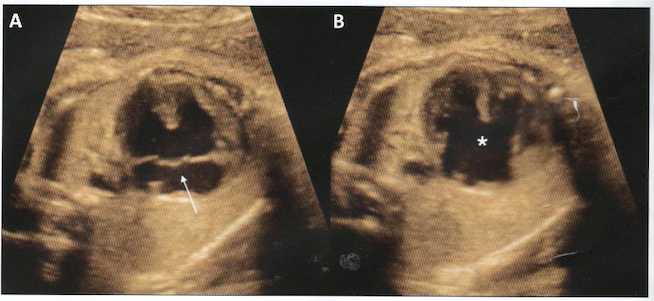
A:systole: valve unique fermée (flèche)
B:diastole: défectueux central majeur (*)
2-Canal atrio-ventriculaire partiel :
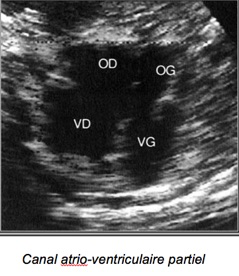
Il existe deux appareils auriculo-ventriculaires distincts.
– Appareil mitral déplacé à l’apex du VG se trouvant ainsi à la même hauteur que la valve tricuspide. –Deux valves alignées.
– S’y associe soit une CIA type ostium primum, une CIV postérieure au contact des valves AV et/ ou une fente de la grande valve mitrale.
Intérêt du codage couleur qui facilite l’identification des communications intra-cardiaques.
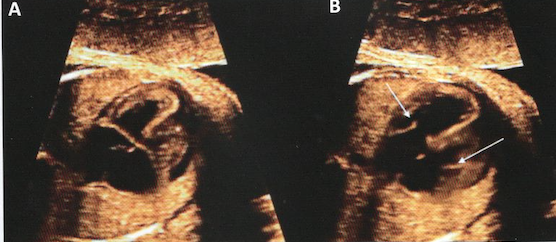
A: systole; B: diastole
(large CIV, petite CIA
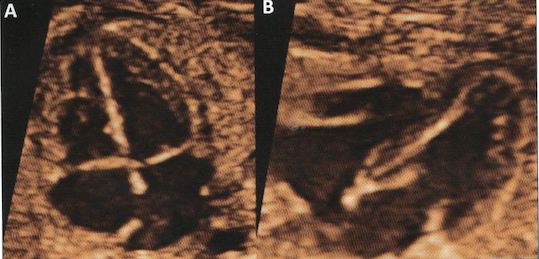
40 % des fœtus trisomique 21 ont une cardiopathie congénitale. Près dune fois sur deux, il s’agira d’un canal atrio-ventriculaire complet.
Anomalies associées :
– bloc auriculo-ventriculaire
– coarctation de l’aorte
– tétralogie de Fallot
– VDDI
– transposition des gros vaisseaux
E-Les dilatations des cavités droites
1-Coarctation de l’aorte
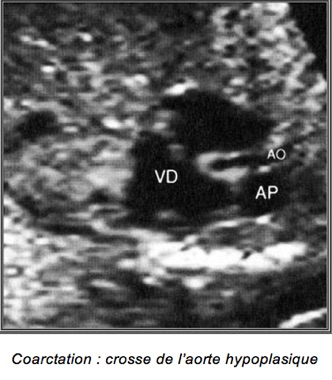
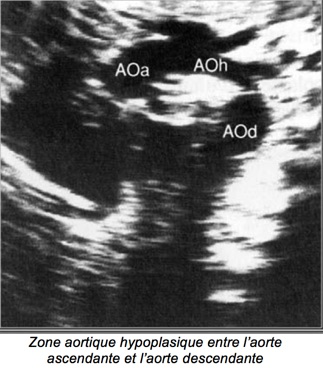
Diagnostic anténatal difficile même en incidence de la crosse de l’aorte.
Elles siègent presque toute au niveau de l’isthme aortique entre l’artère sous-clavière gauche et le canal artériel (alors que les coarctations du grand enfants sont post-ductales).
Soit strictement localisée à l’isthme aortique, soit associée à une hypoplasie plus ou moins longue de la crosse de l’aorte.
Le diagnostic est souvent difficile. Certains signes associés peuvent être évocateurs :
-Crosse de l’aorte de plus en plus hypoplasique avec la grossesse
-Coarctation : crosse de l’aorte hypoplasique
– Dilatation et hypertrophie du VD
– Inégalité du calibre de l’aorte par rapport à celui de l’AP (AP>Ao)
– Insuffisance tricuspide modérée fréquente (due à dilatation VD)
– Souvent bicuspidie aortique ou CIV
Rechercher une anomalie chromosomique (fréquence du syndrome de Turner).
Forme extrême : Interruption de la crosse de l’aorte.
-Toujours CIV associée
– Hypoplasie aorte ascendante et anneau aortique
2-Anomalies cardiaques fonctionnelles (canal artériel restrictif)
Liées à une réduction du calibre du canal artériel :
– soit spontané
– soit après administration d’indométacine (réversible après l’arrêt du traitement)
Dilatation auriculaire et ventriculaire droite + Insuffisance tricuspide
Vitesse du sang au canal artériel normalement < 1,4 m/sec.
F-Les cardiomégalies foetales
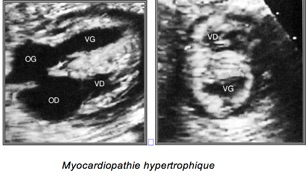
1-Myocardiopathies
Une myocardiopathie est une atteinte primitive du muscle cardiaque qui est anatomiquement normal. On distingue trois types :
-Myocardiopathie dilatée :
– Dilatation et hypokinésie des cavités auriculaires et ventriculaires
– Régurgitation des valves auriculo-ventriculaire
-Myocardiopathie hypertrophique : Augmentation de l’épaisseur des parois ventriculaires et du septum inter-ventriculaire.
-Myocardiopathie restrictive :
– Hyperéchogénicité de l’endocarde
– Diminution de la compliance ventriculaire avec dysfonction diastolique, bas débit et parfois anasarque.
2-Rétrécissement aortique valvulaire
a-Sténose serrée :
-Valves épaissies avec cinétique réduite
– VG dilaté et hypokinétique
b-Sténose modérée:
– Diagnostic échographique difficile
– Parfois, dilatation post-sténotique avec flux accéléré et turbulent dans l’aorte ascendante.
G-Tumeurs du cœur
Souvent histologiquement bénignes, elles peuvent devenir obstructives et avoir un retentissement hémodynamique significatif.
57 % de mortalité in utero.
–Rhabdomyomes :
– Les plus fréquentes (60 % des cas)
– Souvent multiples
– Hyperéchogènes voire calcifiés
– De taille variable
– Des les ventricules ou le septum inter-ventriculaire
– Souvent associés à une sclérose tubéreuse de Bourneville
– Involution post-natale classique
–Fibromes :
– Unique le plus souvent
– Au niveau d’une paroi ventriculaire ou du septum inter-ventriculaire
– Soit hyperéchogène, soit isoéchogène
–Tératomes intra-péricardiques :
– Souvent volumineux
– Masse solide pédonculée
– Parfois responsable d’une compression du cœur
– Hétérogène (alternance de zones hyperéchogènes et kystiques)
– Epanchement péricardique
– Malformations associées (foramen ovale, CIV)
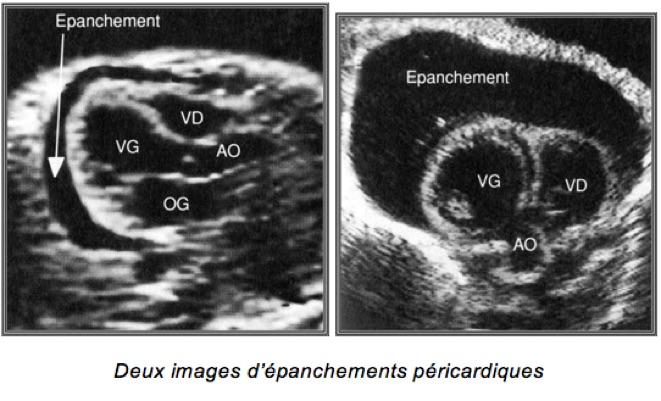
H-Epanchements péricardiques
– Espaces libres d’écho au pourtour des cavités cardiaques
– Parfois premier signe d’une anasarque
– Peut être secondaire à :
• Infection virale
• Anomalie chromosomique
• Tumeur myocardique ou péricardique
III-Malformations conotroncales (non visibles sur la coupe des 4 cavités)
A-Cardiopathies associées à un chevauchement artériel sur le septum interventriculaire
Ces cardiopathies ont en commun :
-CIV haute sous-artérielle, surplombée par un gros vaisseau qui reçoit simultanément du sang des deux ventricules.
-Incidence cinq cavités : Le gros vaisseau chevauche le septum IV et reçoit le sang des deux ventricules, confirmé par le Doppler.
1-LaTétralogie de Fallot
La tétralogie de Fallot est la plus fréquente des cardiopathies congénitales cyanogènes. Elle représente près de 8 % de l’ensemble des cardiopathies congénitales. Son nom est associé à Etienne-Louis Arthur Fallot qui l’a décrite en 1888.
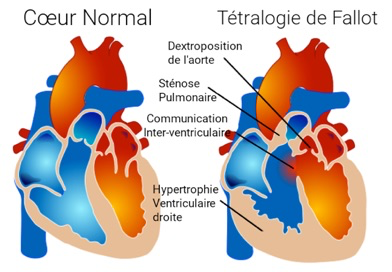
Elle associe :
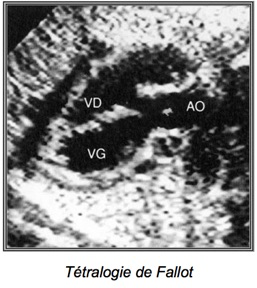
– Chevauchement de l’aorte sur le septum responsable d’une CIV
– Sténose pulmonaire (dont dépend le pronostic)
– Hypertrophie ventriculaire droite (peut manquer in utero)
– Aorte parfois dilatée
– Valves pulmonaires épaissies à mobilité réduite
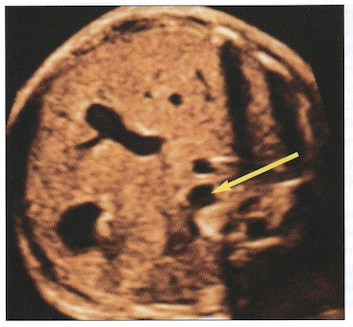
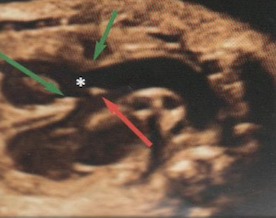
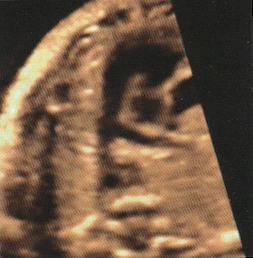
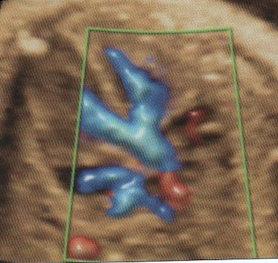
Dans la grande majorité des cas, le traitement, toujours chirurgical, est possible dès la première année de vie et consiste d’emblée en une « réparation complète » de la malformation.
2-Atrésie pulmonaire avec communication interventriculaire (tétralogie de Fallot extrême)
Mêmes caractéristiques que la Tétralogie de Fallot
Mais voie pulmonaire non perméable
Vascularisation pulmonaire assurée soit par le canal artériel, soit par l’intermédiaire de vaisseaux systémiques naissant de la crosse de l’aorte.
Un shunt inversé dans le canal artériel in utero témoigne de l’atrésie pulmonaire.
3-Ventricule droit à double issue (VDDI)
Aorte et AP naissent toutes deux du VD.
4-Tronc artériel commun
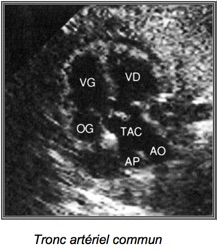
Il résulte d’un défaut de septation du truncus arteriosus qui donne normalement naissance aux deux gros vaisseaux.
A l’échographie on a :
– Un seul gros vaisseau chevauche le septum.
– Il bifurque précocement pour donner naissance à la crosse de l’aorte et à l’AP.
-Valvaes troncales souvent épaissies et fuyantes avec flux diastolique dirigée vers les ventricule.
Dans 50 % des cas : microdélétion 22q11.
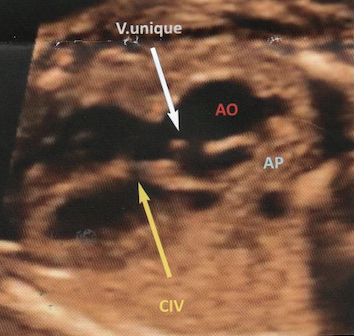
B-Transposition des gros vaisseaux
1-Transposition isolée (D-transposition)
Défaut de rotation du truncus arteriosus
Connexions ventriculo-artérielles anormales :
– L’AP naît du VG
– L’aorte naît du VD et est situé en avant et à droite de l’AP
La survie dépend de la persistance de shunts croisés au niveau du canal artériel et de la cloison inter-auriculaire.
Echographiquement :
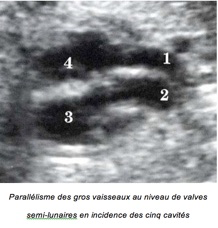
– Parallélisme des gros vaisseaux au niveau de valves semi-lunaires en incidence des cinq cavités
– Aorte antérieure naissant du VD
– AP postérieure naissant du VG et se divisant précocement
– En incidence transversale par les gros vaisseaux, visualisation simultanée des deux gros vaisseaux
– CIV dans 20 % des cas.
2-Transposition congénitalement corrigée (L-transposition)
Double discordance auriculo-ventriculaire et ventriculo-artérielle :
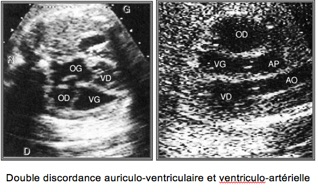
– Ventricule faisant suite à l’OD, de morphologie gauche et se drainant dans l’AP.
– Continuité entre le valve mitrale et la valve pulmonaire.
– Ventricule faisant suite à l’OG, de morphologie droite et se drainant dans l’aorte.
– Les deux ventricules sont l’un à coté de l’autre (alors que normalement le VD est antérieur et le VG postérieur).
La discordance auriculo-ventriculaire est donc corrigée par la discordance venticulo-artérielle.
Ce diagnostic n’est possible que si on a une bonne connaissance des caractéristiques morphologiques des cavités et des gros vaisseaux.
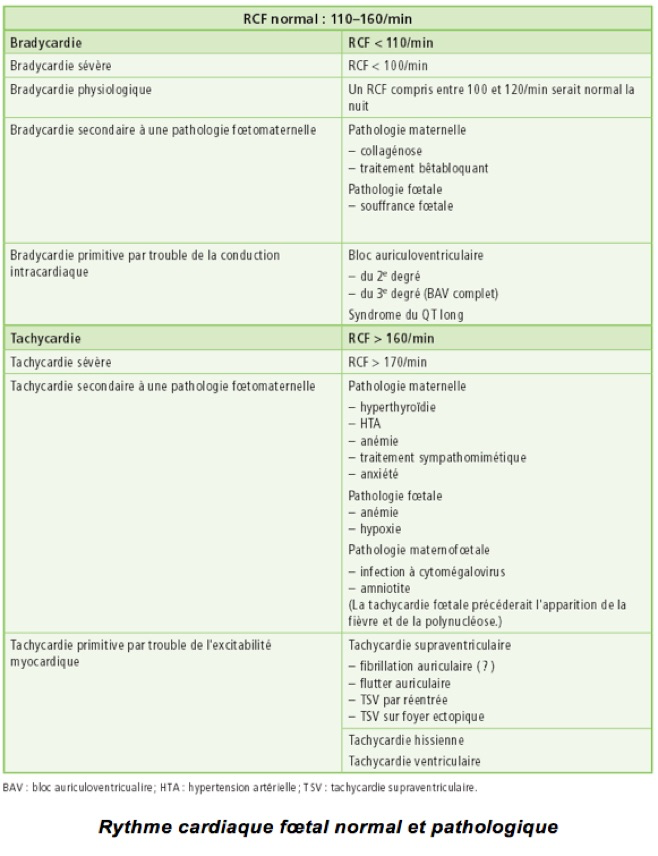
IV-Troubles du rythme cardiaque
Troubles du rythme chez 1 % des fœtus
Normalement : Rythme régulier compris entre 100 et 180 bpm
Le plus souvent bénin, certains troubles du rythme peuvent se compliquer d’une anasarque.
A. Méthodes d’analyse du rythme cardiaque fœtal
1-Echographie mode TM
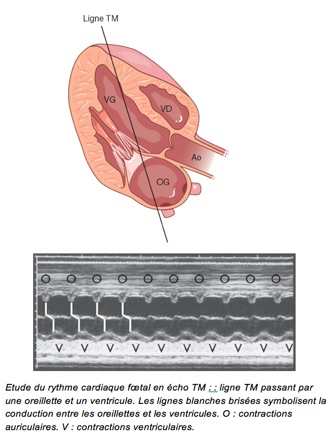
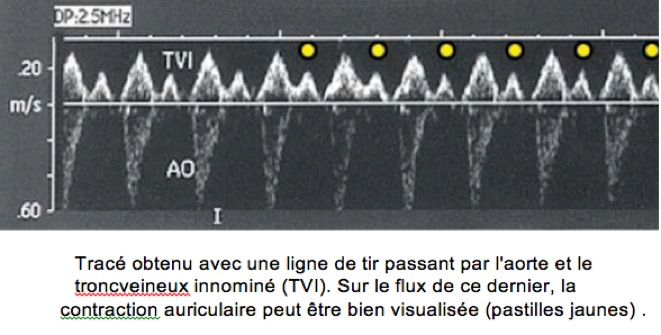
Plan de coupe passant à la fois par une oreillette et un ventricule.
Normalement chaque contraction auriculaire est suivie d’une contraction ventriculaire.
Etude du rythme cardiaque fœtal en écho TM : : ligne TM passant par une oreillette et un ventricule. Les lignes blanches brisées symbolisent la conduction entre les oreillettes et les ventricules. O : contractions auriculaires. V : contractions ventriculaires.
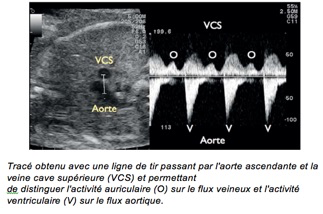
2-Doppler pulsé
Volume d’échantillonnage placé simultanément dans la chambre d’admission et de chasse du VG de façon à enregistrer simultanémentle flux mitral et le flux de la chambre de chasse du ventricule gauche.
B-Extrasystoles
Surviennent le plus souvent sur un cœur sain.
Ce sont des extrasystoles atriales ou ventriculaires ou des épisodes de bloc sino-auriculaire.
C-Tachycardies
Beaucoup plus rares que les extrasystoles, les tachycardies fœtales sont d’une tout autre gravité, car souvent mal tolérées et à l’origine d’une mortalité et d’une morbidité non négligeables, pré- comme postnatale. La possibilité de les traiter efficacement in utero, laissant espérer la naissance
d’un nouveau-né bien portant et normal, en fait un des rares chapitres méritant réellement l’appellation de « médecine fœtale ».
1-Mécanismes et diagnostic
Une tachycardie fœtale peut résulter : d’un trouble de l’excitabilité auriculaire,d’une anomalie de la jonction entre oreillettes et ventricules, à l’origine de phénomène de réentrée ou d’un trouble de l’excitabilité à l’étage ventri-culaire (faisceau de His et myocarde ventriculaire).
2-Troubles de l’excitabilité auriculaire :
fibrillation et flutter auriculaires
a-La fibrillation auriculaire s e r a i t e x c e p t i o n n e l l e chez le fœtus et son existence est même controversée par certains, du moins en l’absence de cardiopathie responsable d’une dilatation importante des oreillettes ou d’anomalie génétique prédisposante, les gènes incriminés étant localisés sur les chromosomes 10q22-24, 6q14-16 et 5p13.
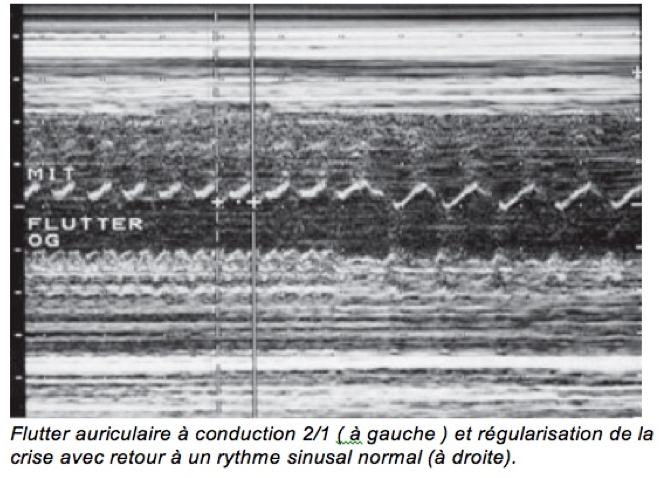
b-Il en va tout autrement du flutter auriculaire, deuxième cause par ordre de fréquence et à l’origine de 20 à 30 % des tachycardies fœtales. Un flutter correspond à un phénomène de « macro-réentrée » activant les oreillettes selon un circuit circulaire rapide, à une fréquence comprise entre 350 et 500/min. À des fréquences aussi élevées, il s’accompagne d’une nette altération de la contraction des oreillettes. Cette activation est transmise aux ventricules, rarement à la même fréquence (conduction 1/1), le plus souvent à une fréquence moindre (120 à 260/min) grâce au blocage de certaines impulsions par le nœud auriculoventriculaire (conduction 2/1 ou 3/1).
On soupçonnera un flutter auriculaire devant une tachycardie découverte au cours du 3ème trimestre et associant :
-une fréquence auriculaire très rapide et régulière ;
-une fréquence ventriculaire moins rapide et le plus souvent irrégulière (bloc de conduction variable). La combinaison la plus évocatrice est celle où la fréquence ventriculaire est régulière et moitié moindreque la fréquence auriculaire. La plus trompeuse est celle où les fréquences auriculaires et ventriculaires, très élevées, sont identiques (conduction 1/1). Il est alors impossible de distinguer le flutter d’une tachycardie par réentrée (voir plus loin). Le dignostic pourra être redressé par l’apparition d’un bloc de conduction sous l’effet du traitement.
3-Tachycardies jonctionnelles par réentrée intranodale ou à la faveur d’une voie accessoire
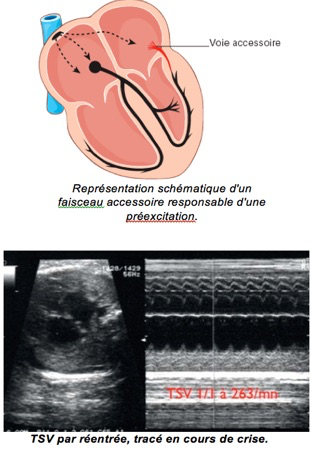
Observées dans 4 à 6 pour 1 000 grossesses, ce sont les tachycardies les plus fréquentes, représentant 60 à 90 % des tachycardies du fœtus. Elles sont secondaires soit à un circuit de « micro-réentrée » se produisant à l’intérieur même du nœud auriculoventriculaire, soit à un circuit de « macro-réentrée » unissant oreillettes et ventricules et empruntant les voies de conduction normales dans un sens et un faisceau accessoire (voie de préexcitation ) dans l’autre Ce dernier mécanisme serait le plus fréquent, impliqué dans plus de 50 % des tachycardies supraventriculaires (TSV) fœtales.
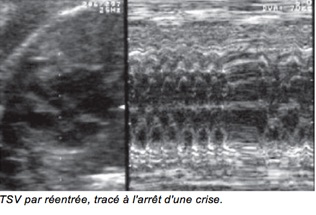
Ces tachycardies par réentrée se caractérisent par :
• une fréquence identique des oreillettes et des ventricules, comprise habituellement entre 220
et 340/min, un peu plus rapide en moyenne que celle observée au cours d’un flutter auriculaire ;
• un début et une fin brusques, la TSV démarrant sur une extrasystole et se terminant par une pause.
D-Bloc auriculo-ventriculaire
Dissociation complète du rythme atriale et du rythme ventriculaire.
Etiologies :
-Malformations cardiaques
– Maladies auto-immunes maternelles avec passage transplacentaire d’anticorps immuns. Impose un bilan immunologique chez la mère (anticorps SSA et SSB).
E-Bradycardies sinusales
Sont dues à une situation de stress maternel ou fœtal, comme un syndrome cave provoqué par le décubitus dorsal.
V-Conduite pratique à tenir devant une cardiopathie
Il y a 5 questions à se poser:
-L’anomalie est-elle isolée?
-Faut-il faire un caryotype?
-Connaitre le pronostic de la malformation
-Définir le lieu de naissance
-Préciser par un conseil génétique le risque de récidive .
VI-Pronostic des cardiopathies
-Celles qui sont à priori curables : CIA, CIV isolées, RVPA
-Celles qui sont en principe incurables : Hypoplasie du VG, cardiopathies univentriculaires, APSO (Atrésie Pulmonaire à Septum ouvert).
-Celles qui sont plus ou moins curables : transposition des gros vaisseaux, tétralogie de Fallot, canal atrio-ventriculaire équilibré, tronc artériel commun, interruption de la crosse aortique, sténose aortique.
VII-Génétique et cardiopathies
13% des cardiopathies sont associées à une anomalie chromosomique. Le risque dépend beaucoup du caractère isolé ou non.
Un caryotype est demandé devant toute cardiopathie, sauf la transposition des gros vaisseaux.
Associations fréqentes :
-Anomalies conotroncales : 50% d’anomalies génétiques associées (délétion 22q1.1, trisomie 21). Il faut donc rechercher une microdélétion 22q11.2 devant toute malformation conotroncale.
-Canal atrio-ventriculaire : 47% (T21, T18, XXX)
-Malposition vasculaire, VDDI : 18%(délétion 22q1.1, Trisomie 21).
-Communication interventriculaire : 16% (trisomies, délétion 22q11, délétion 5).
-Cardiopathie obstructive du cœur gauche : 9% (Turner, trisomie 18, anomalies de structure).
-Ventricule unique et atrésie tricuspide : 8% (trisomie 18).
-Transposition des gros vaisseaux : 0%, donc pas d’étude génétique dans les transpositions isolées confirmées par expert.
-sténose aortique supra-ventriculaire est associée au syndrome de Williams (microdélétion 7q11).
Le syndrome de Di George ou microdélétion 22q11.2
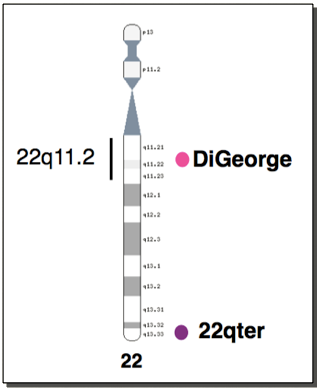
Le syndrome de délétion 22q11.2 est une anomalie chromosomique congénitale, caractérisée le plus souvent par des malformations cardiaques et palatines, une dysmorphie faciale, parfois des anomalies rénales, des malpositions des pieds, hypocalcémie et hypoparathyroï¨dïë¨,¨un retard du développement et une immunodéficience. Cette immunodéficience est un déficit immunitaire primitif provoqué par un développement anormal du thymus qui est hypoplasique. Il s’ensuit une altération du nombre et de la fonction des lymphocytes (essentiellement les lymphocytes T) et dans les suites de la fonction des lymphocytes B (production des immunoglobulines).
Microdélétion 22qter
C’est la suppression de la partie terminale d’un bras long du chromosome 22 provoquant des malformations congénitales multiples en particulier crâniofaciales et des troubles neurologiques.
Le visage est étroit, avec dolicocéphalie, micrognatie, nez proéminent, oreilles dysplasiques, paupières en ptosis et épicanthus. Les orteils 2 et 3 sont en syndactylie. Des malformations cardiaques et rénales peuvent être associées. L’hypotonie est précoce avec un retard de la marche. Une épilepsie est notée dans un tiers des cas. L’acquisition du langage est retardée. Le comportement peut être comparable à celui de l’autisme. La croissance est normale,
L’anomalie qui touche autant les garçons que les filles apparaît de novo : l’examen chromosomique nécessite des examens fins tels que FISH. La délétion est située en 22qter; elle est parfois interstitielle en 22q13,3 respectant le télomèreou en anneau (22r).