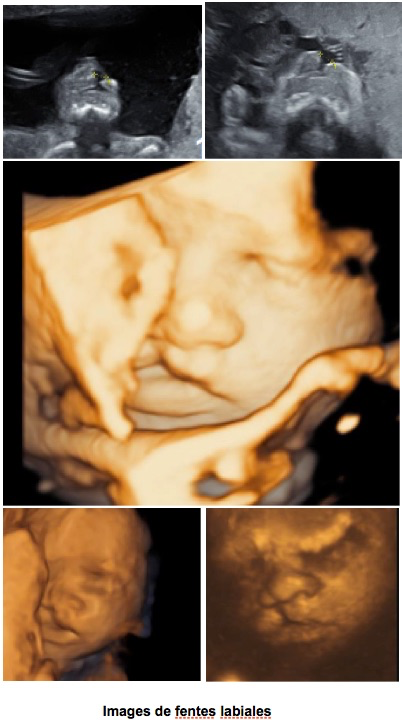
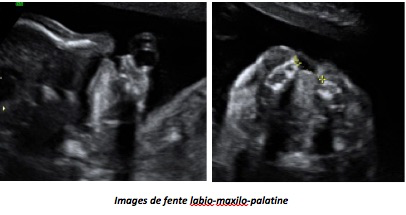
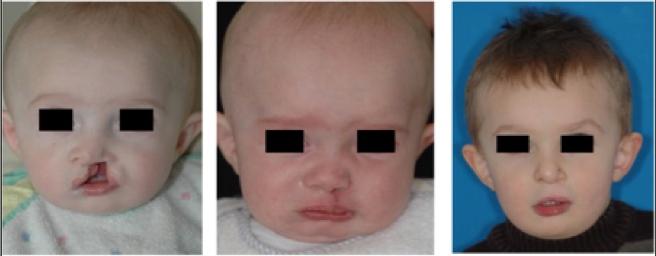
I-Les fentes labio-palatines
La découverte d’une fente conduit donc à rechercher des malformations associées (cérébrales, cardiaque) et à réaliser, le plus souvent, un caryotype pour rechercher une (rare) anomalie chromosomique qui modifierait totalement le pronostic. Diagnostiquées pendant la grossesse, les anomalies labio-palatines isolées bénéficient d’une consultation chirurgicale permettant au chirurgien plasticien d’expliquer aux parents la prise en charge de leur enfant et les résultats de la correction à l’aide d’exemples. Tous ces éléments en facilitent l’acceptation. Les formes plus complexes, polymalformatives, relèvent d’un avis génétique difficile car une même pathologie peut se caractériser par une grande diversité d’expression clinique. Au cours de la grossesse, la tolérance de cette malformation est bonne mais un hydramnios ou un excès de liquide amniotique, par trouble de la déglutition, est habituellement retrouvé dans les larges fentes labio-palatines.
A-Fente labiale uni ou bilatérale
C’est une solution de continuité de la lèvre supérieure sur une coupe frontale, plan d’étude et image de référence.
Une analyse de la lèvre supérieure et de l’arcade dentaire du maxillaire supérieur doit également être obtenue sur une coupe transversale. Elle retrouve une solution de continuité plus ou moins complète, oblique, pouvant se prolonger en arrière vers le palais. Cette coupe transversale est habituellement suffisante pour le dépistage mais la prudence pousse à la recherche systématique de la coupe frontale « de la moustache » car les petites fentes (partielles, du bord de la lèvre) peuvent être « loupées » par une coupe transversale passant plus haut. De même, un profil parfait en 2D peut passer à côté d’une fente labiale.
En cas de fente bilatérale, le bourgeon médian peut réaliser une masse centrale saillante visible sur une coupe sagittale ou transversale. Le profil est nettement perturbé par une sorte de gros bourgeon collé sous le nez, orienté vers l’avant et surplombant la gencive supérieure en retrait.
Une fente médiane est beaucoup plus rare et doit faire évoquer une anomalie cérébrale au niveau de la ligne médiane. Elle peut donc être associée :
-à une malformation nasale : nez petit, une seule narine, voire un proboscis ;
-à un hypotélorisme ;
-à des anomalies oculaires : microphtalmie, cyclopie ;
-et une holoprosencéphalie ;
-tous ces éléments devant faire évoquer l’anomalie chromosomique, surtout la trisomie 13.
B-Fente palatine
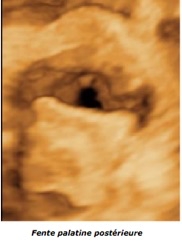
La fente palatine éventuellement associée est beaucoup plus difficile à identifier. Le palais osseux est parfois bien visible sur l’abord du profil ce qui élimine la fente palatine et la présence d’une arcade dentaire complète est rassurante. Mais, inversement, l’image directe de l’ouverture du palais osseux n’est pas évidente, quelque soit la technique.
On s’attachera surtout à rechercher la présence de la langue dans les fosses nasales, signe indirect mais formel, avec patience car ce sont les mouvements musculaires qui la font reconnaître. Sur le profil, il peut exister une ascension de la langue lors de la déglutition, mobile dans la fosse nasale.
II-Anomalies du profil et du nez
A-Hypoplasie des os propres du nez
Les os du nez sont absents ou nettement inférieurs au 5e percentile. Éventuellement associée à un profil plat, l’hypoplasie amène immédiatement à rechercher une trisomie 21. L’hypoplasie peut aussi être un signe d’appel d’une dysplasie squelettique rare telle que la dysostose cléido-crânienne où il existe une aplasie ou hypoplasie claviculaire.
B-Ensellure nasale marquée
Ce signe est habituellement décrit dans l’achondroplasie , le nanisme thanatophore et le syndrome d’Apert. L’appréciation est assez subjective et il existe des variations interindividuelles à ce niveau. Cependant, les formes pathologiques sont souvent caricaturales.
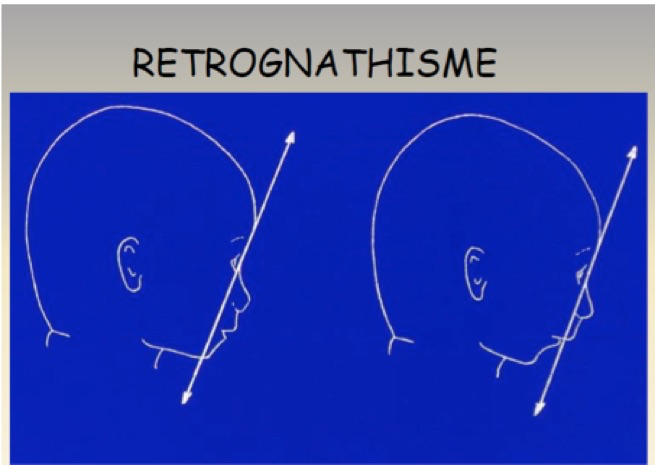
C-Rétrognathisme
Il se caractérise par le recul du menton en arrière du plan tangent à la partie inférieure de l’os frontal. Sur une coupe frontale de la face (coupe « de la moustache »), on ne parvient pas à visualiser le menton dans le même plan que le nez et la bouche. Le rétrognathisme est caricatural et caractéristique du syndrome de Pierre Robin mais on le rencontre aussi dans le syndrome de Cornelia de Lange, dans la trisomie 18, la dysostose cranio-faciale.
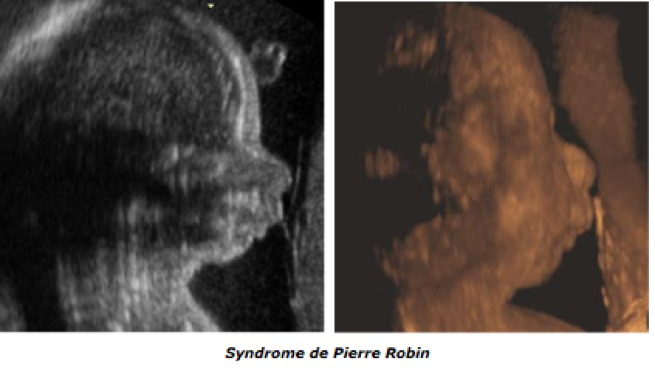
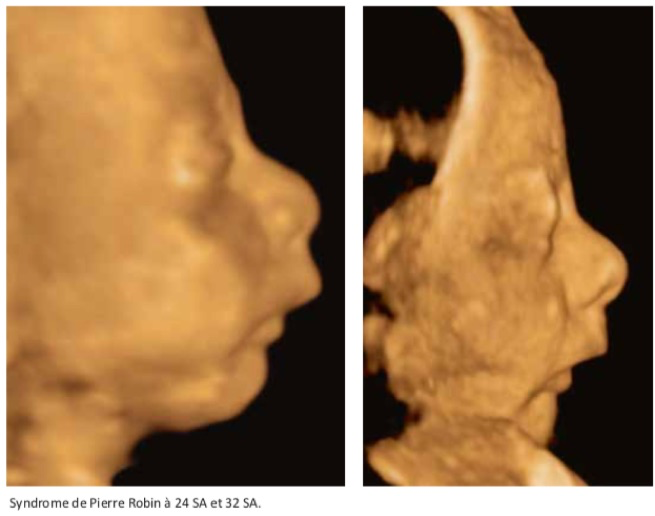
La syndrome de Pierre Robin (on parle pluotôt de la «séquence de Pierre Robin») associe dans sa forme complète un microrétrognathisme, une fente palatine, une glossoptose et un hydramnios par troubles de la déglutition. Elle peut être isolée, d’origine tératogène (alcool, valproate), ou chromosomique (trisomie 13, 9, 18 et 21), mais, deux fois sur trois, elle s’intègre dans un syndrome polymalformatif : syndrome de Strickler, syndrome de Treacher Collins, syndrome vélo-cardio-facial, un syndrome de Nager, une dysplasie osseuse. La glossoptose risque d’entraîner une asphyxie néonatale ce qui nécessite une prise en charge adaptée à la naissance.

D-Prognathisme du maxillaire inférieur
Il est en général relatif et secondaire à une hypoplasie maxillaire supérieure qu’on peut retrouver dans le syndrome de Pfeiffer (craniosynostose avec brachycéphalie et gros orteil large en abduction.
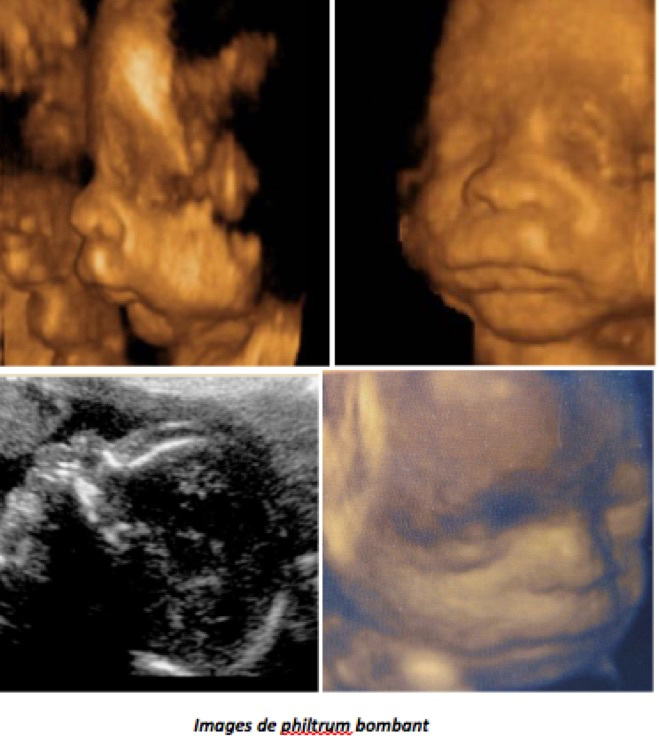
E-Philtrum court
Il est assez caractéristique des formes sévères de syndrome d’alcoolisation fœtale (SAF), avec le contexte et souvent un RCIU, son appréciation est aussi très subjective d’autant que le faciès SAF est parfois simplement familial, en dehors de toute intoxication. On trouve aussi une lèvre supérieure courte et bombée dans le syndrome de Noonan.
F-Tuméfaction des parties molles
Elle peut correspondre à un hémangiome un hamartome, une méningo-encéphalocèle.
G-Tératome naso-pharyngé
On parle d’ épignathus. Très rare, environ 1/30 000 naissances, ce tératome multitissulaire (comme le tératome sacro-coccygien) prend volontiers naissance au niveau du palais, de l’ethmoïde ou du sphénoïde et s’extériorise par la bouche ou le nez, déformant le profil de façon variable selon le volume mais souvent monstrueuse. La tumeur est homogène ou hétérogène (tissu osseux), elle distend et efface la bouche et/ou le nez et s’accompagne souvent d’un hydramnios par trouble de la déglutition. Des fentes faciales ou des anomalies cérébrales peuvent s’y associer ainsi que des malformations cardiaques.
H-Absence de nez (arhinie)
Elle s’observe dans le cadre d’une holoprosencéphalie. Absence totale avec souvent présence d’un bourgeon au-dessus du plan orbitaire correspondant au proboscis, en cas de cyclopie et d’ethmocéphalie, ou pyramide rudimentaire avec une seule narine.
III-Macroglossie
En échographie la macroglossie est évoquée devant l’interposition constante de la langue entre les arcades dentaires ou la protrusion persistante de la langue hors de la bouche même au cours des mouvements buccaux. Elle s’observe dans le syndrome de Beckwith-Wiedemann dans lequel lui sont associées une viscéromégalie (foie et rein) et une omphalocèle. À la naissance, l’enfant peut présenter une hypoglycémie. Une protrusion linguale peut également être observée chez 20 % des fœtus trisomiques 21 après 28 SA.
IV-Tumeurs buccales et linguales
Peuvent exister : kyste lingual, le kyste sublingual, le lymphangiome kystique lingual, le tératome buccal.
V-Anomalies oculo-orbitaires
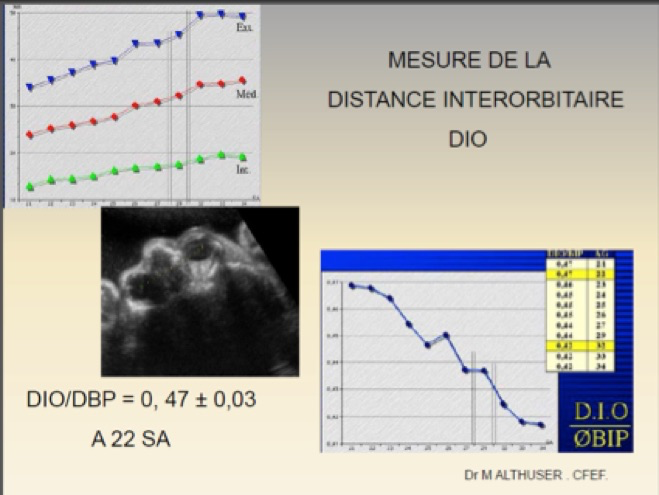
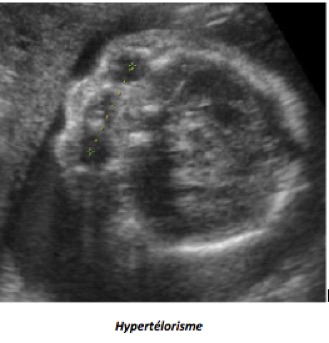
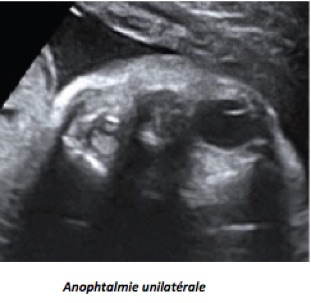
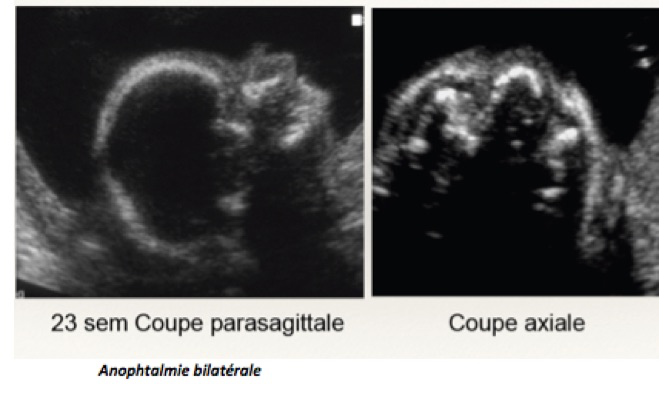
Les malformations oculaires et orbitaires sont rares et généralement retrouvées lors du bilan d’un syndrome polymalformatif et/ ou chromosomique. On peut observer : anomalies des orbites, anophtalmie, microphtalmie, hypertélorisme, hypotélorisme, cyclopie, masses orbitaires ou péri-orbitaires, cataracte.
VI-Anomalies de l’oreille
La biométrie et la morphologie du pavillon de l’oreille peuvent être étudiées lors de l’échographie de 22 SA. Au moins une oreille devrait être vue. Son analyse doit être systématique en cas d’anomalie faciale.
Le mode 3D permet une étude plus facile de la position et de la morphologie de l’oreille. La découverte de tubercules à localisation prétragienne est possible.
En pratique on distingue :
-les aplasies de l’oreille externe qui peuvent être unilatérales dans le syndrome de Franceschetti, le syndrome de Goldenhar ou les dystrophies mandibulofaciales ;
-les hypoplasies souvent bilatérales dans le syndrome de Treacher-Collins (Franceschetti-Klein), la maladie de Crouzon, les dystrophies mandibulofaciales, certaines aneuploïdies.
