I-Les anomalies de taille
A-La microcéphalie
Elle se définit par un diamètre bipariétal et une circonférence céphalique inférieurs au 3e percentile de l’âge gestationnel considéré.
La microcéphalie traduit le ralentissement ou l’arrêt de l’expansion encéphalique du fait de:
-malformations diverses : holoprosencéphalie, céphalocèle, troubles de la migration avec ventriculomégalie, dysgénésie du corps calleux et lissencéphalie le plus souvent dans le cadre d’un syndrome polymalformatif (lissencéphalie de type I et type III, syndrome de Smith-Lemli-Opitz, syndrome de Dubowitz… et ostéochondrodysplasies) ;
-aberrations chromosomiques fréquentes : trisomie 9, 13, 22 et diverses délétions (délétion 4p ou syndrome de Wolf-Hirschhorn) ;
-microcephalia vera par un arrêt précoce de la migration et de la prolifération neuronales s’associant à une lissencéphalie et une hypoplasie du corps calleux, alors que dans le radial microbrain la morphologie est normale ;
-causes infectieuses ou métaboliques : CMV, rubéole, herpès, alcool, cocaïne, phénylcétonurie…
En cas de lésions « clastiques » précoces et importantes (aprosencéphalie, atélencéphalie) on peut observer une craniosténose globale secondaire ou même un chevauchement des sutures.
B-La macrocéphalie
Elle se définit par un BIP et une une circonférence céphalique supérieurs au 97e percentile, en discordance avec le reste de la biométrie, ce qui exclut une erreur de datation ou une macrosomie fœtale. La macrocéphalie peut être secondaire à l’expansion de l’encéphale ou s’intégrer à une anomalie squelettique (ostéochondrodysplasie, dysostose) ou un syndrome polymalformatif. Ainsi:
-hydrocéphalie de différentes origines ;
-hydranencéphalie par destruction ischémique ;
-troubles de la migration neuronale :
-syndrome de Walker-Warburg : lissencéphalie de type II, hydrocéphalie, hétérotopie cérébelleuse, ± syndrome de Dandy-Walker, céphalocèle (et décollement rétinien),
-hémimégalencéphalie avec ventricules plus ou moins dilatés et déformés : syndromes neuro-cutanés (Protée, Klippel-Trenaunay, neurofibromatose de type I…),
-mégalencéphalie : dilatation ventriculaire, corps calleux quelquefois épaissi, dysplasie corticale et operculation incomplète,
-overgrowth syndromes (Sotos, Weaver),
-anomalies métaboliques rares (et souvent postnatales).
II-Les anomalies des ontours du crâne
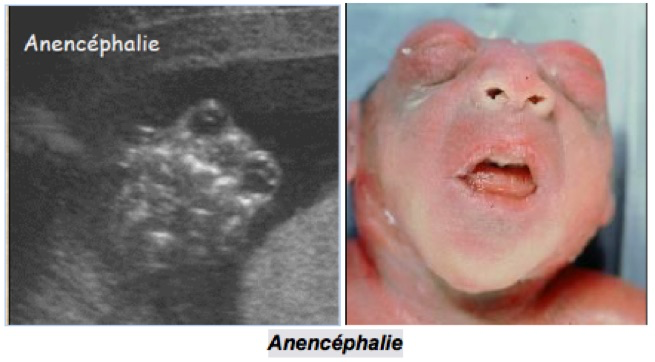
A-L’anencéphalie
est une malformation du système nerveux central qui découle de l’absence de la fermeture normale du tube neural à l’extrémité antérieure, généralement entre le 23e et le 26e jour de la grossesse. Cette malformation cause l’absence partielle ou totale de l’encéphale, du crâne, et du cuir chevelu.
Rarement d’origine génétique, elle peut être secondaire à un diabète insulino-dépendant mal équilibré ou à une carence en acide folique (ce qui justifie la prévention par la prise d’acide folique : 0,4 mg/j, deux mois avant si possible et deux mois après la conception).
En échographie, le diagnostic est possible dès 12-13 SA et se caractérise par un pôle céphalique anormal (structure de l’encéphale inhabituelle, absence de ligne interhémisphérique) recouvert parfois par une membrane méningée. Au deuxième trimestre, les signes en sont :
-pas de voûte crânienne visible ;
-BIP impossible à mesurer ;
-base du crâne et face identifiables ;
-yeux paraissant exorbités ;
-rachischisis associé dans 25 % des cas ;
-fente labio-palatine associée dans 10 % des cas ;
-malformations urinaires (16 %), digestives (6 %), cardiaque (4 %) ;
-hydramnios fréquent ;
-mouvements fœtaux paraissant saccadés et amples provoqués par le contact du moignon céphalique avec l’utérus.
B-Céphalocèle
C’est une hernie du contenu encéphalique à travers une brèche de la voûte ou de la base du crâne donnant une lacune de la voûte avec protrusion d’une poche méningée (méningocèle) pouvant contenir du tissu cérébral (encéphalocèle).
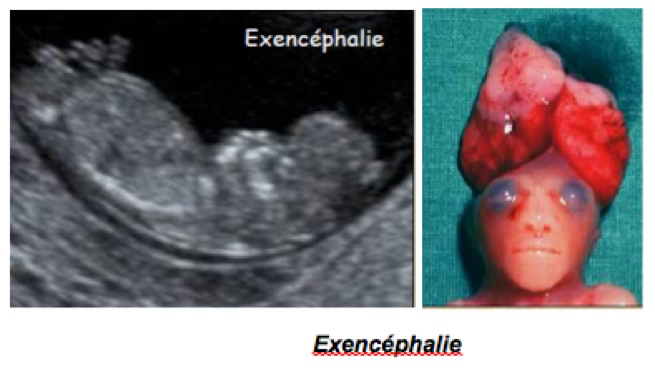
De nombreux syndromes sont associés à une céphalocèle, en particulier le syndrome de Meckel-Gruber (autosomique récessif) comportant une céphalocèle occipitale, une dysplasie rénale multikystque, une hexadactylie et un anamnios.
L’iniencéphalie associe une céphalocèle occipitale, un rachischisis des premières vertèbres cervicales responsable d’une hyperextension du cou.
L’encéphalocèle se retrouve dans les formes majeures de maladie des brides amniotiques où l’encéphale anormal est adhérent au placenta, fixé lors des mouvements foetaux. Dans cette forme on retrouve une dysmorphie faciale complexe à type de fentes faciales asymétriques et obliques, d’hypertélorisme, d’anomalie de l’ensellure nasale, de microphtalmie ou anophtalmie unilatérale.
C-Les images d’addition de la voûte cranienne
1-hémangiome : C’est une masse finement échogène, aux contours bosselés, bien individualisée par le Doppler couleur.
2-Lipome : Il peut provenir d’une localisation méningée ou atteindre le corps calleux (lipome frontal). De contours réguliers, il est peu échogène.
3-Kyste épidermoïde : il est caractérisé par un aspect liquidien et des contours nets.
E-Les déformations des contours du crâne
1-Les diagnostics de dolichocéphalie (petit BIP, mais circonférence céphalique normale) et de brachycéphalie (BIP élevé mais circonférence céphalique normale) ne sont pas pathologiques. Une dolichocéphalie est fréquemment notée chez un fœtus en présentation du siège. Une brachycéphalie isolée correspond le plus souvent à une variante ethnique sans signification pathologique.
2-Défauts de minéralisation. Une voûte crânienne se déformant facilement à l’appui de la sonde n’est pas toujours le témoin d’une pathologie. Mais, associée à une trop bonne définition cérébrale sous jacente, on peut évoquer un défaut de minéralisation, en recherchant d’autres signes échographiques :
-une hypophosphatasie (autosomique récessif) : crâne légèrement élargi, ventricules normaux, thorax étroit, absence d’ossification des côtes et vertèbres, raccourcissement de tous les membres et importante déminéralisation ;
-une ostéogénèse imparfaite de type II (autosomique dominant): macrocrânie, platispondylie, thorax étroit et cals osseux des côtes, membres inférieurs courts, incurvés, hypominéralisés, fracturés ;
-une dysostose cleido-crânienne (autosomique dominant) : brachycéphalie, thorax étroit, hypoplasie des clavicules, membres normaux…
3-Les craniosténoses
La plus commune (la moitié des cas) est la soudure prématurée de la suture sagittale suivie de celle de la suture coronale. De très nombreux syndromes sporadiques ou génétiques ont été décrits.
Le diagnostic est rarement posé avant vingt semaines d’aménorrhée et sera aidé par l’échographie 3D-4D. On peut individualiser plus particulièrement :
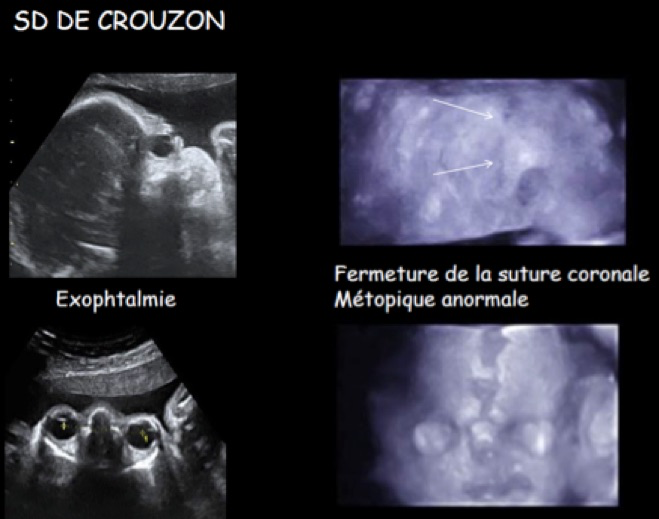
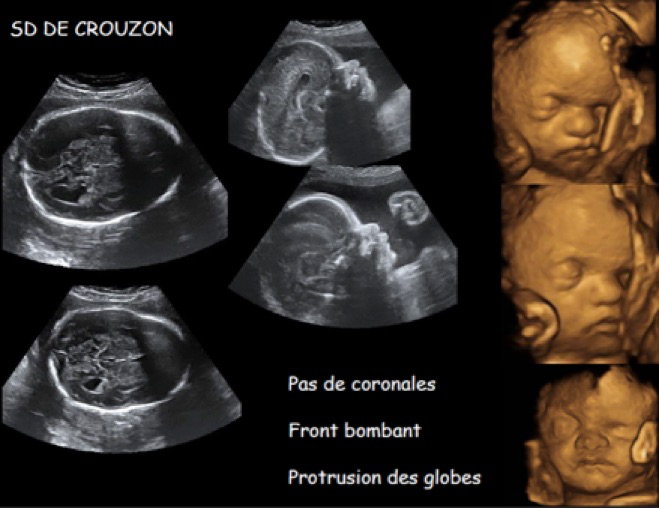
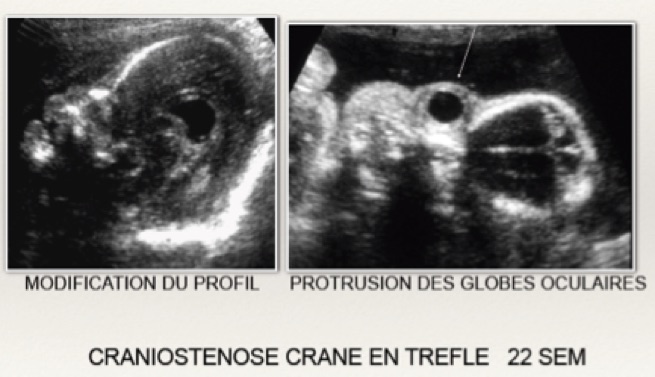
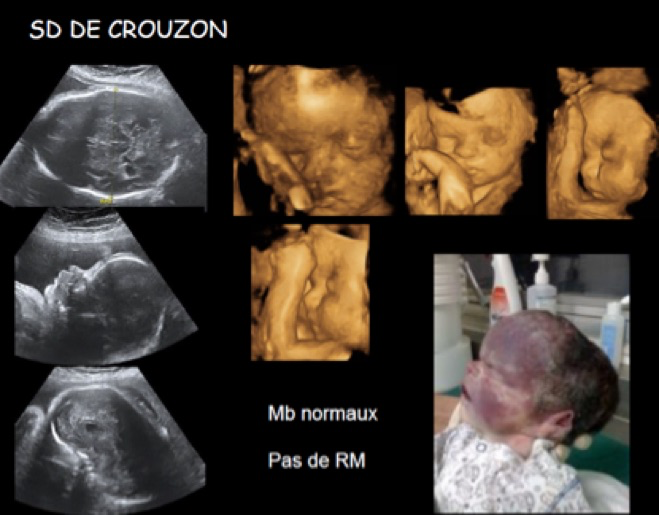
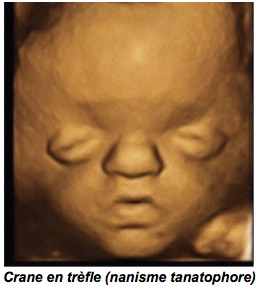
-un crâne en forme de trèfle avec exophtalmie, hypertélorisme, oreilles bas implantées, hypoplasie maxillaire et prognathisme mandibulaire mais sans anomalie des extrémités qui évoque un syndrome de Crouzon (1/ 25 000 naissances) ;
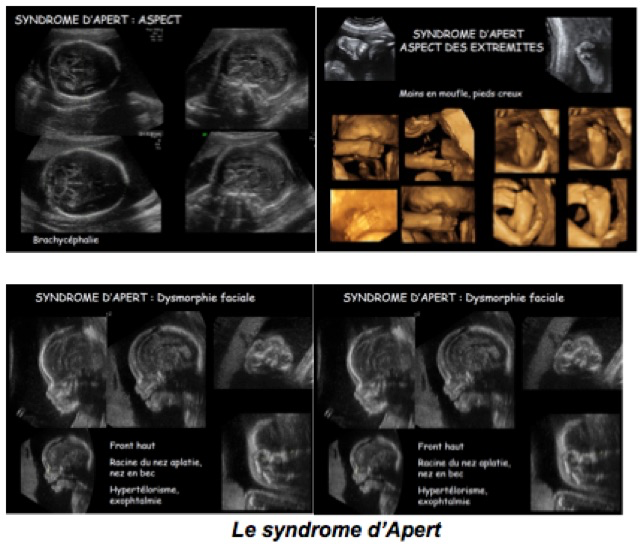
-L’acrocéphalosyndactylie ou syndrome d’Apert (1/70 000 naissances) caractérisée par une brachycéphalie symétrique avec front verticalisé (turricéphalie), hypertélorisme et mains « en moufles » peut également être évoquée. Il peut s’y associer des malformations cérébrales diverses : agénésie du corps calleux, céphalocèle, hydrocéphalie, mégalencéphalie, exophtalmie et hypertélorisme.
-Le syndrome de Pfeiffer (très rare) caractérisé par une brachycéphalie et une acrocéphalie asymétrique, une dysmorphie faciale (visage aplati avec hypoplasie maxillaire et prognathisme relatif, oreilles bas implantées, racine du nez déprimée, exophtalmie et hypertélorisme). Les extrémités sont anormales avec une déviation en varus du gros orteil, des orteils larges, une syndactylie partielle du deuxième et troisième doigt avec aspect trapézoïde du pouce, une syndactylie partielle du deuxième et troisième doigt de pied. Il peut s’y associer une synostose du coude, une hydrocéphalie et une atrésie des choanes responsable d’hydramnios.
Rappelons enfin qu’un aplatissement et un rétrécissement bitemporal (feston frontal ou signe du « citron ») est fortement évocateur, au deuxième trimestre, d’une dysraphie ouverte, avec dans ce cas, une ventriculomégalie habituellement modérée et une petite fosse postérieure (malformation de Chiari II).

III-Les dilatations ventriculaires ou ventriculomégalies
A-Technique de mesure
La mesure d’un ventricule latéral se calcule au niveau du carrefour sur une coupe transversale transthalamique du cerveau. Elle se prend perpendiculairement à la paroi ventriculaire, du bord interne au bord externe de la corne occipitale du ventricule latéral, immédiatement en arrière du plexus choroïde en évitant soigneusement d’intégrer la substance blanche hypoéchogène. Le ventricule latéral le plus éloigné de la sonde est bien vu, alors que le plus proche est difficilement accessible. Son étude nécessite la réalisation de coupes coronales postérieures. La mesure moyenne d’un carrefour ventriculaire est de 6,1 mm (± 1,3 mm). Elle ne devrait pas dépasser 10 mm quel que soit l’âge de la grossesse. Néanmoins une mesure à 10,5 mm reste à la limite de la physiologie chez un garçon, surtout s’il est macrosome. Elle serait plus inquiétante chez une fille de poids moyen. Une asymétrie ventriculaire (différence supérieure à 2,5 mm) est parfois notée. Elle n’est pas inquiétante s’il n’y a pas de dilatation ventriculaire.
Le diamètre du troisième ventricule mesuré sur une coupe transversale ne doit pas dépasser 3,5 mm et celui du quatrième ventricule 4,8 mm.
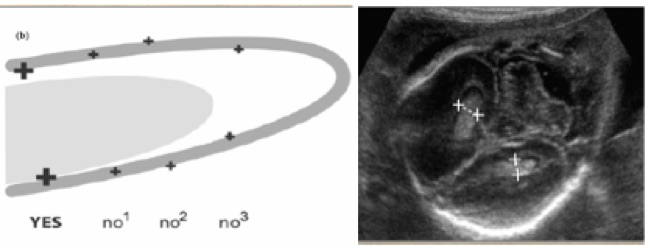
B-Degrés de gravité
-de 10 à 12 mm, la dilatation est modérée, elle est à surveiller mais elle est rarement pathologique ;
-de 13 à 15 mm, la dilation est moyenne et nécessite d’emblée des explorations complémentaires;
-au-delà de 15 mm, le pronostic est très réservé. La céphalométrie est variable. En cas de microcéphalie, une ventriculomégalie importante traduit une importante réduction de volume du parenchyme cérébral;
-ce n’est souvent qu’à partir de 20 à 25 mm de dilatation des carrefours que l’on notera une augmentation significative de la céphalométrie avec un cortex cérébral aminci en périphérie;
-enfin, dans les formes majeures on peut retrouver une fenestration septale avec passage d’un plexus choroïde dans la cavité ventriculaire controlatérale voire une asymétrie marquée dans la dilatation des ventricules.
C-Conduite à tenir
Il faut rechercher des malformations extracérébrales rendant le pronostic défavorable et chercher la cause la plus fréquente par l’étude du rachis à la recherche d’une myélodysraphie et l’étude du corps calleux. Il faut demander les examens complémentaires indispensables en cas de doute étiologique après avoir repris l’étude des antécédents médicaux et du déroulement de la grossesse : échographie de référence, IRM, examen cytogénétique (liquide amniotique ou mieux sang fœtal), bilan infectieux maternel et fœtal (CMV, toxoplasmose, herpès), recherche d’incompatibilité plaquettaire, test de Kleihauer. Il faut proposer une surveillance échographique toutes les 2 à 4 semaines dans les ventriculomégalies modérées et moyennes isolées pour lesquelles le diagnostic étiologique est revenu négatif, et une IRM de contrôle vers 30-32 SA (étude de la giration et du parenchyme cérébral) pour toutes les dilatations > à 12 mm et les formes évolutives.
D-Les causes des ventriculomégalies
On distingue, les « ventriculomégalies », acquises par destruction ainsi que malformatives, où la pression du LCS est normale dans le ventricule, et les « hydrocéphalies » où l’augmentation résulte d’une hyperpression intraventriculaire.
1-Ventriculomégalies acquises par destruction
Elles sont d’origines infectieuses (CMV, Toxoplasmose), hémorragiques ou hypoxiques. On les suspecte devant les signes suivants : parois ventriculaires échogènes de façon diffuse ou disséminée traduisant une épendymite, un ou plusieurs kystes sous-épendymaires, pouvant atteindre 10 mm de diamètre, à paroi fine, localisés en particulier au niveau du sillon thalamo-caudé ou en regard de la tête du noyau caudé, échos diffus ou regroupés en amas correspondant à des débris, caillots, brides dans le contenu ventriculaire, échogénicité excessive des plexus choroïdes, aspect du tissu avoisinnant (nécrose, hémorragie).
2-Ventriculomégalies acquises malformatives
a-Dysgénésie du corps calleux
Cela va de l’agénésie totale ou partielle à l’hypoplasie globale ou segmentaire. Elle affecte 0,3 % de la population générale et 2 à 3 % de la population neuro-psychiatrique. La récurrence est faible en dehors de cas à transmission génétique. Les anomalies associées sont très fréquentes (50 %) et responsables en grande partie de la gravité du pronostic.
-Signes échographiques directs : absence d’individualisation du corps calleux sur une coupe sagittale médiane ou coronale antérieure . Cependant, si une agénésie totale est de diagnostic simple, une agénésie partielle, voire une hypotrophie calleuse est de diagnostic plus difficile. On pourra aussi vérifier l’absence de gyrus cingulaire et l’absence de visibilité en Doppler couleur de l’artère péricalleuse.
-Signes échographiques indirects : sur une coupe transversale : absence de cavum du septum lucidum mais Il peut être présent dans l’agénésie partielle postérieure, dilatation modérée, bilatérale et symétrique des cornes occipitales (colpocéphalie), déplacement en dedans des parois ventriculaires occipitales qui tendent à devenir parallèles à la scissure interhémisphérique), l’aspect en triple feuillet de l’écho médian constitué par la faux du cerveau et les faces internes des hémisphères cérébraux, cornes frontales, effilées à leurs extrémités, petites, éloignées l’une de l’autre, élargissement de la scissure interhémisphérique surtout frontale, ascension et la dilatation du 3e ventricule avec kyste interhémisphérique possible. Et sur une coupe sagittale (mais plus tardivement à partir de la fin du deuxième trimestre en raison de la giration) on constate une désorganisation de l’architecture cérébrale en regard du versant médial des ventricules latéraux donnant une disposition radiaire des sillons. Cette constatation est surtout vraie au niveau du genou et du corps car, en regard des lobes occipitaux, les sillons sont normalement perpendiculaires au corps calleux
b-Agénésie partielle du corps calleux
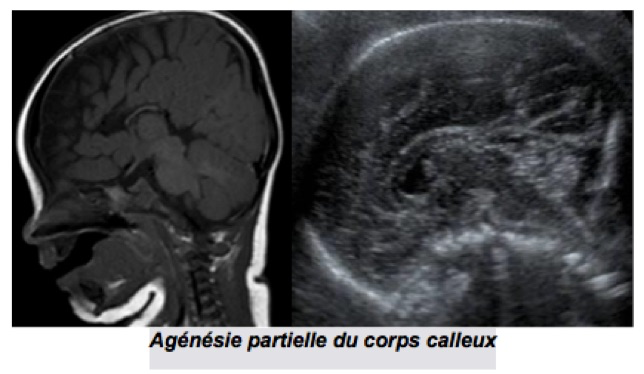
Causes:
-Acquises : infectieuses, nutritionnelles, métaboliques, toxiques, vasculaires
-Génétiques (s’intégrant dans de nombreux syndromes), expliquant les anomalies du système nerveux central fréquemment associées (holoprosencéphalie, Chiari II, schizencéphalie, dysplasie septo-optique…) ; chez une fille, il faut évoquer un syndrome d’Aicardi où le retard mental est sévère, s’il existe des anomalies costales et vertébrales à type d’hémivertèbres ou de fusions vertébrales
-Secondaires à un obstacle au développement par un lipome et/ou un kyste interhémisphérique
c-Holoprosencéphalie
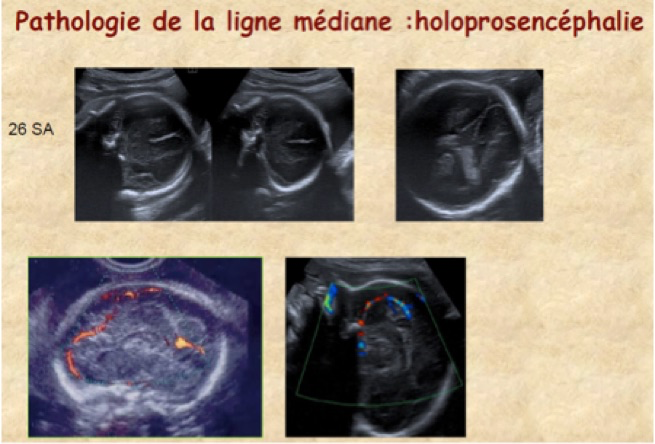
L’absence ou le clivage incomplet du cerveau antérieur responsable d’anomalies du développement intracrânien et/ou de la face. L’holoprosencéphalie est classée en quatre types selon l’importance des lésions (et le pronostic) : alobaire, semi-lobaire, lobaire, syntélencéphalie (fusion hémisphérique médiane qui ne peut être diagnostiquée en échographie anténatale). Dans 50 % des cas, l’origine est chromosomique impliquant les chromosomes 2, 7, 13, 18 et 21. Certains sites chromosomiques associés avec une holoprosencéphalie sont connus. Il faut noter une plus grande fréquence chez les patientes diabétiques insulinodépendantes, dans le syndrome d’alcoolisation fœtale, dans le syndrome de Smith-Lemli-Opitz.
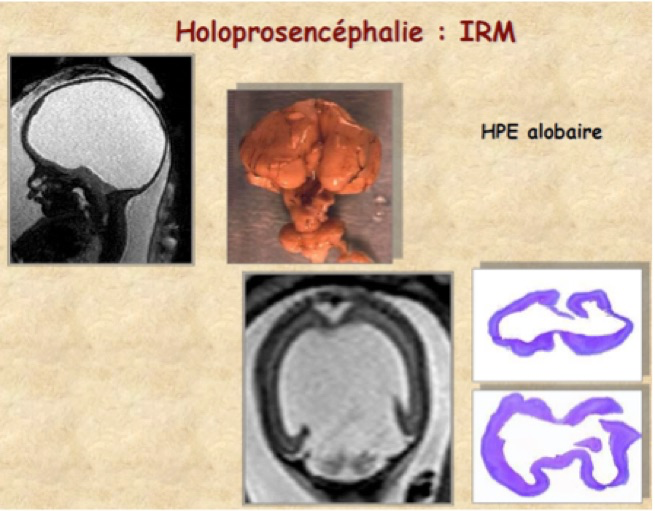
L’échographie de l’holoprosencéphalie alobaire montre :
-une large cavité ventriculaire unique à concavitédes thalamus accolés par leurs faces internes ;
-l’absence de corps calleux, du cavum du septum pellucidum, de faux et de scissure interhémisphérique
-une microcéphalie ;
-un tissu cérébral, fusionné au niveau de la ligne médiane, plaqué en avant ;
-une hypoplasie de la fosse cérébrale postérieure ;
-de très fréquentes anomalies de la face : hypotélorisme, agénésie maxillaire, fente labiale ou labio-palatine, arhinencéphalie, proboscis, cyclopie.. Celles-ci permettent le diagnostic différentiel avec une hydrocéphalie majeure (où les thalamus ne sont pas accolés) et une hydranencéphalie (où persiste souvent un peu de la faux du cerveau).
-accessoirement, un trajet aberrant de l’artère cérébrale antérieure en coupe sagittale, refoulée en avant vers la table osseuse ce qui en allonge le trajet horizontal et en réduit le calibre.
L’échographie de l’holoprosencéphalie semi lobaire montre:
-un ventricule unique arciforme se prolongeant en arrière par des cornes occipitales séparées et dilatées ;
-un accolement partiel des thalamus séparés par un troisième ventricule rudimentaire ;
-une scissure interhémisphérique à la partie postérieure du cerveau ;
-une fosse cérébrale postérieure quasi normale ;
-l’absence de cavum du septum pellucidum ;
-l’absence du genou et du corps du corps calleux, mais splénium présent ;
-des anomalies fréquentes de la face mais moins graves que dans la forme alobaire avec surtout un hypotélorisme presque constant.
L’holoprosencéphalie lobaire est de diagnostic échographique plus difficile voire impossible en anténatal car la scissure interhémisphérique est presque complète à l’exception de la région antérieure où les deux lobes frontaux sont réunis à leur base. Elle peut s’intégrer dans une dysplasie septo-optique. Les cornes frontales sont fusionnées. Il n’existe pas de septum ou de cavum du septum pellucidum. Le genou du corps calleux est absent alors que sa partie postérieure est présente. Parfois le corps calleux est présent mais il est alors hypotrophique et fin, exceptionnellement normal. Le V3 est formé. Les piliers antérieurs du trigone sont fusionnés.
d-Anomalies de la migration neuronale
Elles accompagnent un grand nombre d’anomalies cérébrales acquises ou de nature syndromique.
– Les hétérotopies sont fréquentes, mais ne sont que très rarement diagnostiquées en échographie anténatale. Elles se situent le plus souvent en région sous-épendymaire se traduisant alors par une paroi ventriculaire épaissie, échogène et festonnée avec une discrète dilatation ventriculaire. Des nodules d’hétérotopies plus volumineux , hyperéchogènes, sont parfois retrouvés en région sous corticale. Elles peuvent accompagner des dysplasies corticales, lissencéphalie et schizencéphalie.
– Les (hémi-)mégalencéphalies traduisent des troubles de la migration et de la prolifération neuronales dans divers syndromes (Protée, lipomatose encéphalo-cranio-cutanée, syndrome de Klippel-Trenaunay…).
– Les anomalies de la giration sont vues plus tardivement après 30 SA et sont difficiles à diagnostiquer si elles sont isolées. Dans un contexte de microcéphalie ou de dilatation ventriculaire il est utile de rechercher les scissures accessibles précocement en échographie : scissure sylvienne dès 18 SA, scissure pariéto-occipitale et scissure calcarine dès 22-24 SA. Les lissencéphalies peuvent être identifiées in utero avec, en général, une ventriculomégalie, une dysgénésie du corps calleux, un aspect en sablier des contours du cerveau secondaire à un défaut d’operculation sylvienne ainsi que dans le type I (syndrome de Miller-Dieker , de Norman-Robert), une microcéphalie et une dysmorphie faciale, dans le type II (syndrome de Walker-Warburg, de Fukuyama), une macrocéphalie, une hypoplasie de la fosse cérébrale postérieure, parfois céphalocèle ou syndrome de Dandy-Walker et dans le type III (syndrome de Neu-Laxova), une microcéphalie avec également atteinte cérébelleuse et médullaire engendrant un syndrome d’akinésie fœtale.
3-Ventriculomégalies par Hydrocéphalie
L’hydrocéphalie se définit par une distension ventriculaire secondaire à une augmentation du volume de LCS sans préjuger de sa cause. Le périmètre crânien est alors augmenté.
Les étiologies sont :
-une hyperproduction de LCS sécrété par un papillome des plexus choroïdes. Une forme unilatérale est possible ;
– un défaut de résorption suite à un processus infectieux, hémorragique ou une hyperpression veineuse par un anévrysme de la veine de Galien. Des anomalies de la voûte et de la base du crâne peuvent entraîner un trouble de résorption et une compression des sinus veineux dans l’ostéochondrodysplasie et la craniosténose ;
-une obstruction du transit du liquide cérébro spinal :
-au niveau des foramens interventriculaires, le plus souvent par une épendymite d’origine infectieuse ou hémorragique, parfois une compression par la tumeur d’une sclérose tubéreuse (tuber pouvant dégénérer) ou par un kyste suprasellaire. Cette obstruction peut être unilatérale ;
-au niveau de l’aqueduc du mésencéphale (aqueduc de Sylvius) par un kyste arachnoïdien ou par une sténose que l’on peut rencontrer dans l’hydrocéphalie liée à l’X (garçon, hydrocéphalie triventriculaire, pouce en adduction) ;
-au niveau de la fosse cérébrale postérieure, la compression peut être secondaire à un kyste arachnoïdien, à un syndrome de Chiari II, de Dandy-Walker, à un rhombencéphalosynapsis.
IV-Images liquidiennes anormales
A-L’Hydranencéphalie
L’hydranencéphalie correspond (dans sa forme majeure et bilatérale) à la destruction totale des hémisphères cérébraux remplacés par une vaste cavité liquidienne entourée des méninges. Elle est secondaire à une thrombose des carotides internes elle-même secondaire à une infection (toxoplasmose, CMV), une malformation vasculaire ou des troubles hémodynamiques. Elle peut être exceptionnellement unilatérale.
L’échographie montre :
-un aspect plus ou moins anéchogène de la cavité crânienne avec au début un contenu grumeleux présentant un niveau de sédimentation, puis une structure purement liquidienne par la suite ;
– la présence de la faux du cerveau qui n’est parfois visible que partiellement ;
– la persistance de reliquats cérébraux en région frontale et occipitale vascularisés par les artères cérébrales antérieures et postérieures et leurs anastomoses ;
– la fosse cérébrale postérieure, la tente du cervelet, les noyaux de la base et le tronc cérébral sont préservés ;
– parfois, la voûte crânienne peut être déformée avec macrocéphalie par une production continue de LCS ;
– la mobilité fœtale est conservée.
B-La porencéphalie
Au sein du parenchyme cérébral apparaissent des cavités remplies de LCS, secondairement à une destruction parenchymateuse par des lésions hypoxo-ischémiques localisées, d’origine embolique ou infectieuse. Elles siègent le plus souvent dans le territoire de l’artère cérébrale moyenne.
On retrouve en échographie :
une zone intraparenchymateuse, parfois volumineuse, unique ou multiple, hypo ou anéchogène, aux contours mal limités, à paroi fine, pouvant augmenter de volume ;
– une communication possible avec le ventricule et les espaces sous arachnoïdiens ;
– une expansion ventriculaire en regard ;
– l’absence d’effet de masse sur les structures avoisinantes.
Dans le cadre d’une grossesse gémellaire monozygote avec mort d’un fœtus, une porencéphalie disséminée (encéphalopathie multikystique ou leucomalacie périventriculaire) chez le fœtus survivant est une complication redoutée.
C-La schizencéphalie
Il s’agit d’une « fente » hémisphérique généralement périsylvienne secondaire à une hypoxie cérébrale avant dix semaines d’aménorrhée, entraînant une destruction segmentaire de la matrice germinale qui aboutit à une absence de migration neuronale. Une origine génétique est possible. Elle peut être unique ou bilatérale et, en fonction de la largeur de la fente, on parle de schizencéphalie de type I (bords accolés) ou de type II (bords ouverts). Une évolution du type I vers le type II est possible. Le pronostic neurologique est réservé s’il s’agit d’une schizencéphalie de type II bilatérale, meilleur s’il s’agit d’un type I unilatéral qui s’accompagne toutefois souvent de dysplasie corticale controlatérale. Des anomalies locomotrices sont plus fréquentes si la schizencéphalie concerne le lobe frontal.
En échographie, la fente du type I est difficile à diagnostiquer et pourrait être évoquée devant l’absence inexpliquée de cavum du septum pellucidum. C’est l’IRM qui confirmera le diagnostic.
En faveur du type II, on retiendra : la présence d’une fente, à bords écartés mettant en communication le ventricule latéral et les espaces sous arachnoïdiens périphériques en région sylvienne, les bords de la fente soulignés par la substance grise dysplasique, le cavum du septum pellucidum non vu et une absence de corps calleux en regard de la fente, une ventriculomégalie plus ou moins importante.
D-L’anévrysme de la veine de galien
Malformation rare, ne représentant que 1 % des malformations artério-veineuses cérébrales congénitales, l’anévrysme de la veine de Galien correspond à une dilatation pseudo-anévrysmale de la veine de Galien. Celle-ci met en relation normalement le système veineux profond avec le sinus droit et le torcular (confluent des sinus veineux). Il existe dans cette pathologie une communication anormale par fistules artério-veineuses avec des branches artérielles d’origine carotidienne ou vertébro-basilaire.
L’utilisation du Doppler couleur facilite le diagnostic anténatal devant :
-une collection anéchogène, paramédiane, de forme oblongue, site en arrière et au-dessus des thala
– un flux turbulent en Doppler couleur ;
– une ventriculomégalie possible, modérée par compression de l’aqueduc de Sylvius ou défaut de résorption par augmentation de la pression veineuse, une encéphalomalacie et une atrophie cérébrale ;
– une dilatation des cavités cardiaques droites avec régurgitation tricuspidienne ;
– une dilatation des vaisseaux jugulaires ;
– des signes d’insuffisance cardiaque droite tels qu’une hépatomégalie, un épanchement des séreuses, un hydramnios ;
– rarement ou plus tardivement une anasarque fœto-placentaire.
Le pronostic est souvent mauvais avec une mortalité néonatale précoce de 50 % par décompensation hémodynamique et un risque élevé de séquelles neurologiques. Pour les 75 % de nouveau-nés survivants, traités par embolisation, l’évolution neurologique est habituellement normale.
E-Le Kyste arachnoïdien
Formation bénigne remplie de liquide céphalorachidien, le kyste se développe partout où l’on trouve du tissu arachnoïdien avec comme localisations préférentielles, la fosse cérébrale moyenne, la région suprasellaire, la citerne quadrijumelle, la région interhémisphérique et la fosse cérébrale postérieure. Il ne présente pas de communication avec le système ventriculaire mais peut communiquer avec les espaces sous arachnoïdiens. L’évolution anténatale du volume kystique est variable : stabilité le plus souvent, régression spontanée, ou à l’inverse aggravation rapide en fin de grossesse. Le pronostic est fonction de la localisation du kyste, de son volume et de la présence de rares malformations associées comme une dysgénésie du corps calleux dans les formes inter hémisphériques.
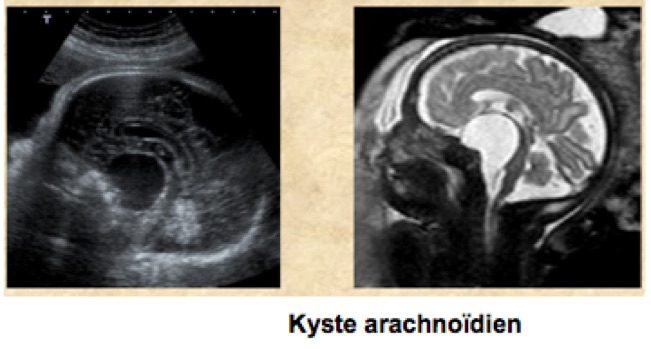
Signes échographiques :
-une masse anéchogène, de volume variable ;
-généralement unique mais pouvant être polylobée ;
-à contours bien définis et réguliers ;
-entraînant une compression des structures de voisinage ;
-pouvant s’étendre jusqu’à la périphérie, sans interposition parenchymateuse ;
-ne communiquant pas avec les ventricules latéraux.
Le diagnostic différentiel se pose essentiellement avec une porencéphalie ou un kyste épendymaire, mais également avec un exceptionnel hématome par hémorragie méningée se situant dans l’espace sous-arachnoïdien ou sous-dural , hypoéchogène et périphérique refoulant le parenchyme cérébral en dedans et pouvant le comprimer. L’hématome ensuite devient hétérogène puis volontiers échogène si un ou plusieurs caillots se constituent.
F-Les pseudo-kystes épendymaires
Non bordés par de l’épendyme, ces pseudo-kystes se développent sous celui-ci, dans la paroi ventriculaire à l’endroit où persiste un peu de matrice germinale. Ils apparaissent comme de multiples formations anéchogènes ou cloisonnées, uni ou bilatérales, de petite taille, situés sous les cornes frontales, au niveau de la région thalamo-caudée, ou à l’angle externe des ventricules latéraux, développés dans la paroi ventriculaire mais indépendants de la lumière ventriculaire et également du parenchyme cérébral.
Le pronostic est bon si la germinolyse est isolée, mais beaucoup plus réservé lorsqu’elle est secondaire à une infection type CMV, Toxoplasmose, rubéole, herpès ou à une hémorragie sous-épendymaire, ou à une anomalie chromosomique, ou un épisode hypoxo-ischémique par hémorragie materno-fœtale massive, hypotension, intoxication au CO, drogue, jumeaux transfuseur-transfusé, déficit en pyruvate-carboxylase, déficit en holocarboxylase synthétase ou encore un syndrome cérébro-hépato-rénal de Zellweger (maladie génétique rare, 1/50 000 naissances, secondaire à une anomalie structurelle des péroxysomes, appartenant aux leucodystrophies et affectant la croissance de la gaine de myéline et dont l’évolution est généralement mortelle dans les six premiers mois de vie) associant une dysmorphie faciale à type d’hypertélorisme et d’épicanthus, des anomalies oculaires (glaucome et cataracte), des kystes du cortex rénal, des anomalies de la giration (pachy et polymicrogyrie), une hépatomégalie par fibrose et stéatose hépatique.
G-Les kystes épendymaires
Ils se développent dans le tissu cérébral où il peuvent être volumineux présentant de multiples logettes avec des contours réguliers, facilement différenciés d’une porencéphalie, rencontré en particulier dans le syndrome oro-facio-digital de type I. Ils se développent aussi dans les espaces sous arachnoïdiens, en périphérie ou en interhémisphérique où il peuv être unique et petit ou multiples en grappes pouvant atteindre la voûte crânienne sans interposition de parenchyme cérébral. Ces kystes ne sont pas communicants entre eux. La recherche de lésions sous-jacentes en IRM est proposée.
H-Les (pseudo) kystes des plexus choroïdes
Pseudo kystes car il s’agit de formations liquidiennes dans les plexus chroroïdes mais sans paroi propre. Ces collections sont fréquentes (3 %) dans la période 16-19 SA qui correspond au pic de production du LCS. En l’absence de toute autre anomalie associée, elles sont physiologiques quelque soit l’aspect échographique. La régression spontanée est observée dans 80 % des cas après 26 SA. Elle est constante à terme.
Il faut rechercher des anomalies morphologiques ou biométriques associées. Leur présence serait alors un signe d’appel important en faveur d’une aneuploïdie (6 %) particulièrement d’une trisomie 18. Dans cette hypothèse, on analysera attentivement, le cœur, la face et le profil, les mains, et on cherchera un retard de croissance intra-utérin (constant) et/ ou un hydramnios. En l’absence d’anomalies échographiques, un contrôle ultérieur de sécurité à quinze jours peut être pertinent.
I-Anomalies du cavum du septum lucidum
Délimité latéralement par les cornes frontales ventriculaires, au dessus par le corps calleux, au dessous par le fornix, il s’étend de la lamina terminalis au splénium. Il est un élément essentiel à visualiser lors de l’échographie du pôle céphalique. Il commence à s’effacer à partir de 34 SA.
Un cavum trop large ets souvent normal mais il faut aussi évoquer une anomalie du développement cérébral et du système limbique (trisomie 18…).
L’absence de cavum se rencontre avant tout dans l’agénésie du corps calleux mais aussi dans l’agénésie septale acquise.
V-Les anomalies tissulaires
A-Les calcifications
Elles peuvent être la conséquence d’un syndrome infectieux fœtal type toxoplasmose ou cytomégalovirus mais également d’une hémorragie intracrânienne focalisée ou intraventriculaire.
B-Lipome intracrânien
C’est une formation hyperéchogène et homogène, aux contours bien définis , soit arrondie en regard du genou du corps calleux qui présente une dysgénésie, et fréquemment associée à des anomalies dysraphiques en particulier frontales : céphalocèle, defect frontal et lipome frontal témoignant d’un développement précoce , soit allongée tout le long du corps calleux, pouvant entourer le splénium sans retentissement sur le corps calleux, ni autre anomalie, témoignant ainsi d’un développement plus tardif . Le Doppler couleur montre l’artère péricalleuse incluse dans le lipome.
C-tératome intracrânien
Les aspects échographiques sont :
– une formation multikystique, hétérogène à composante hyperéchogène, parfois calcifiée, responsable de déformations et de déviations des structures encéphaliques ; au maximum, aucune structure normale n’est individualisable ;
-une macrocrânie, avec hydrocéphalie symétrique ou non ;
– une croissance très rapide avec effraction possible de la voûte ;
– un hydramnios fréquent.
V-Les anomalies de la fosse postérieure
A-Mega-grande citerne
La profondeur de la grande citerne mesurée sur une coupe transversale entre soit le sommet d’un hémisphère, soit le bord externe du vermis cerébelleux et le bord interne de l’écaille occipitale varie de 2 à 10 mm, avec une moyenne à 6 mm pour la deuxième méthode. Son augmentation doit être considérée comme une variante de la normale en l’absence d’association malformative anatomique ou biométrique du cervelet. Elle n’entraîne pas de dilatation ventriculaire ou d’effet de masse sur le cervelet car elle communique librement avec les espaces sous-arachnoïdiens péri-médullaires.
La présence possible d’éléments linéaires médians (simples, doubles ou multiples) tendus entre le vermis et l’occiput correspond à des trabéculations normales de l’arachnoïde et à la faux du cervelet.
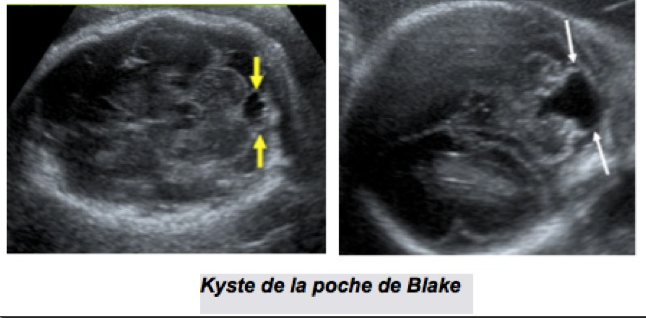
B-Kyste de la poche de Blake
La poche de Blake (sus et rétrocérebelleuse) est secondaire à un retard de l’ouverture de la toile choroïdienne. Exceptionnellement cette poche peut constituer un kyste fermé de la grande citerne formé par une évagination caudale de la toile choroïdienne. Cette formation kystique postéro-inférieure entraîne une bascule et une horizontalisation du vermis créant une ouverture inférieure du V4. Une hydrocéphalie sus-jacente par compression est possible, le kyste ne communiquant pas avec les espaces sous-arachnoïdiens péri-médullaires.
C-Kyste arachnoïdien rétrocérebelleux
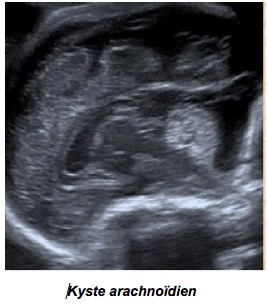
Médian ou latéralisé, à contours nets et géométriques, il est anéchogène, parfois volumineux avec effet de masse surélevant la tente du cervelet, déformant les hémisphères cérébelleux (mais sans les comprimer réellement) et pouvant entraîner une hydrocéphalie. Le pronostic est généralement bon.
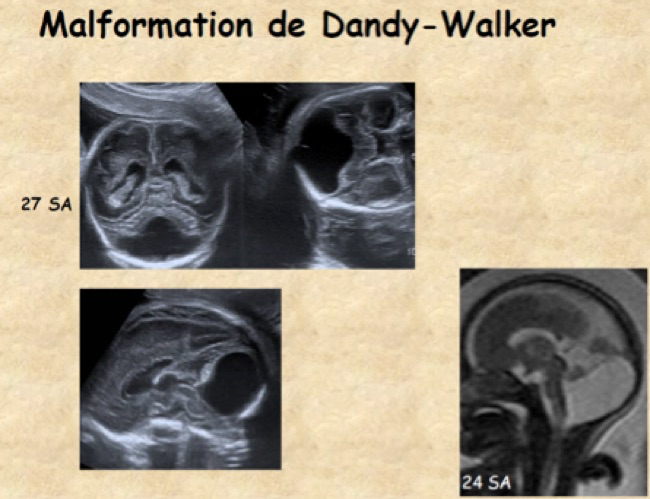
D-Syndrome de Dandy-Walker
Le syndrome de Dandy-Walker (ou malformation de Dandy-Walker) associe une dilatation kystique du V4, une agénésie complète ou partielle du vermis et une hypoplasie des hémisphères cérébelleux responsables de la distension de la fosse cérébrale postérieure.
Il représente 14 % des formations kystiques de la fosse cérébrale postérieure et touche 1/30 000 naissances. 40 % des enfants décèdent la première année, le retard psychomoteur est fréquent chez les autres et il est corrélé au degré d’atteinte du vermis cérébelleux. Le plus souvent isolé avec une récurrence de 1 à 5 %, il est associé dans 35 % des cas à des anomalies chromosomiques. Il est présent également dans de nombreux syndromes polymalformatifs génétiques ou sporadiques.
L’échographie associe à des degrés divers:
-Elargissement de la fosse postérieure par la dilatation kystique du V4, triangulaire à base externe.
– Absence de faux du cervelet
– Surélévation de la tente du cervelet.
– Hernie sus-tentorielle possible dans la citerne quadrigéminale
– Hypogénésie ou agénésie (25 %) du vermis .
– Déplacement latéral et antérieur des hémisphères cérébelleux qui sont plus ou moins hypoplasiques, de façon symétrique ou non.
– Désorganisation et hétérotopie cérébelleuse.
- Tronc cérébral hypoplasique.
Les malformations associées sont nombreuses au premier rang desquelles on retrouve les anomalies cardiaques (38 %). On retrouve aussi : dysmorphie faciale et fente palatine (26 %), dysraphie, poly et syndactylie (28 %), malformations génito-urinaires (28 %) et digestives.
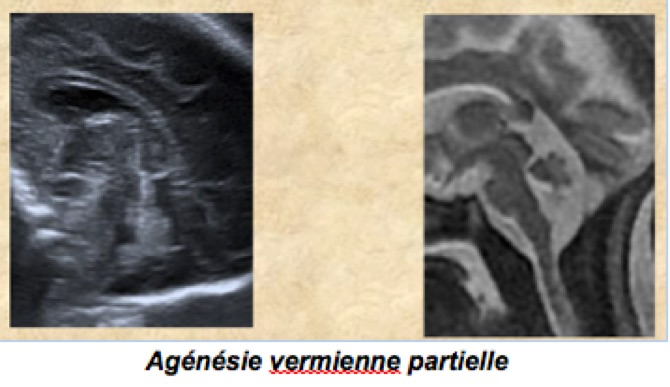
E-Aplasies ou hypoplasies vermiennes
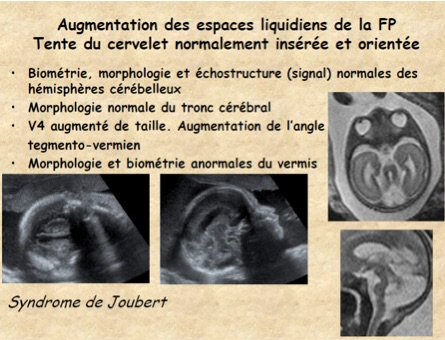
1-Syndrome de Joubert
C’est une maladie autosomique récessive rare, d’étiologie inconnue et de pronostic péjoratif (décès avant trois ans).
En échographie, on note :
-une aplasie vermienne totale ou partielle entraînant une déformation du V4 en aile de chauve-souris ou triangulaire et une communication large du V4 avec la grande citerne qui peut être augmentée de volume -une hypoplasie du tronc cérébral et des pédoncules cérébelleux supérieurs;
-des anomalies de la migration et de la giration ;
-une surélévation de la tente du cervelet ;
-une céphalocèle occipitale (25 %), une agénésie du corps calleux, une hydrocéphalie.
L’association avec une polykystose rénale (7 %), une poly ou une syndactylie (10 %) est évocatrice. À noter qu’il existe une dystrophie rétinienne dans 50 % des cas.
2-Syndromes polymalformatifs
Devant une aplasie ou hypoplasie vermienne, de multiples syndromes polymalformatifs peuvent être suspectés en échographie :
-avec une aplasie vermienne constante dans les syndromes de Debakan, de Walker-Warburg, cérébro-oculo-musculaire…
-l’aplasie vermienne est en revanche inconstante dans les syndromes d’Ellis-Van Creveld, de Smith-Lemli-Opitz, de Cornelia de Lange, de Goldenhar, de Meckel-Gruber, oro-facio-digital de type II et III, CHARGE association.
F-Aplasies ou hypoplasies des hémisphères cérébelleux
1-Aplasie de l’ensemble du cervelet
Elle est très rare, avec hypoplasie du tronc et résidus du toit du V4 et des pédoncules, souvent associée à d’autres anomalies cérébrales.
2-Hypoplasie globale symétrique
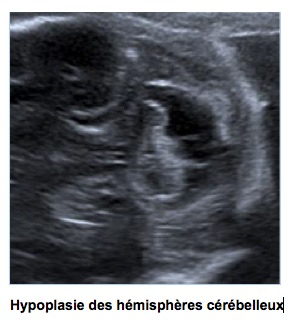
Elle donne plus ou moins un aspect de méga-grande citerne avec une largeur du cervelet faible. Elle est secondaire à l’alcoolisme, au CMV, à une trisomie 21 ou 18, ou se rencontre dans de nombreux syndromes dont certains vont poursuivre la dégénérescence cérébelleuse en postnatal. Il existe de fréquentes associations à d’autres malformations sus-tentorielles ou de la ligne médiane.
3-Hypoplasie unilatérale
Le vermis et l’hémisphère controlatéral y participent plus ou moins. Elle est d’origine infectieuse ou ischémique.
G-Anomalies vasculaires
1-Angiome télangiectasique du rhombencéphale
Il s’agit d’une malformation vasculaire capillaire rare se développant dans le rhombencéphale et se traduisant en échographie comme une lésion cérébelleuse très échogène, sans modification des structures anatomiques de la fosse cérébrale postérieure, sans vascularisation décelable au Doppler et sans caractère d’évolutivité. Cette absence d’évolution la différencie d’une lésion hémorragique ou ischémique que l’on pourrait évoquer et qui évoluerait vers une atrophie cérébelleuse. Le pronostic de cet angiome, généralement découvert de façon fortuite, est bon.
2-Autres anomalies vasculaire
La thrombose d’un ou de plusieurs sinus veineux cérébraux crée une dilatation veineuse sus-jacente. En fosse cérébrale postérieure le thrombus s’individualise par une masse échogène arrondie avec au Doppler couleur une interruption du flux sanguin au niveau des vaisseaux dilatés.
Une structure anormale échogène et liquidienne de la fosse cérébrale postérieure, latéralisée et créant un effet de masse vers l’avant peut évoquer également un hématome de la fosse cérébrale postérieure ou un hématome sous-dural. Dans cette seconde hypothèse une disparition complète des images anté-natales confirmée par l’imagerie postnatale est possible et le pronostic neurologique est bon. Les facteurs maternels responsables d’une hémorragie fœtale doivent être recherchés en particulier une thrombocytopénie alloimmune idiopathique, la maladie de Willebrand, un traumatisme abdominal… Comme facteur causal chez le fœtus on peut citer le syndrome transfuseur-transfusé, la mort fœtale d’un jumeau en cas de gémellaire monochoriale, une hémorragie fœto-maternelle…
H-Petite fosse postérieure
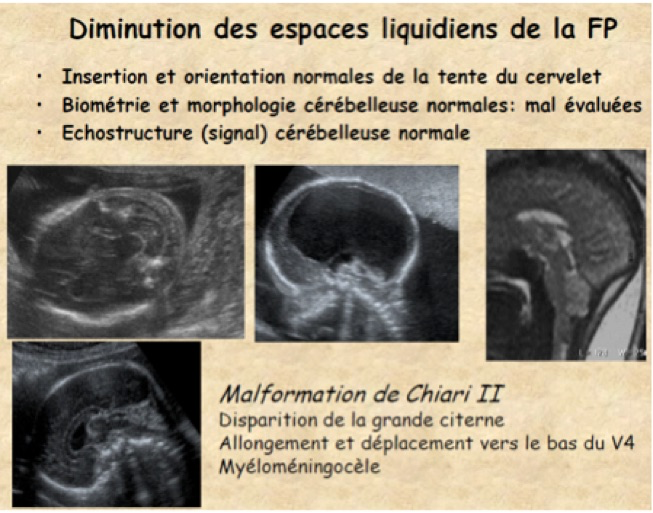
1-Chiari II
Cette malformation est secondaire à un defect du tube neural avec fuite du liquide céphalorachidien responsable notamment d’un défaut de distension de la vésicule rhombencéphalique perturbant l’induction du mésenchyme avoisinant et du chondrocrâne avec petite fosse postérieure. Le tronc et le cervelet se développent dans cette petite fosse induisant une hernie au travers des foramens magnum et ovale. Des modifications sus-tentorielles découlent de ces anomalies.
En échographie, à l’étage sous-tentoriel on note :
-une grande citerne inexistante ;
-un vermis inférieur ectopique ;
-des hémisphères cérebelleux attirés vers le bas formant une incurvation à concavité antérieure se moulant sur le tronc cérébral (signe de la banane) ;
-un V4 aplati, étiré ;
-une horizontalisation de la tente du cervelet sur une coupe frontale postérieure.
À l’étage sus-tentoriel on retrouve :
-un feston frontal (signe du citron ;
-une ventriculomégalie ;
-souvent une agénésie partielle du corps calleux ;
-des troubles de la migration.
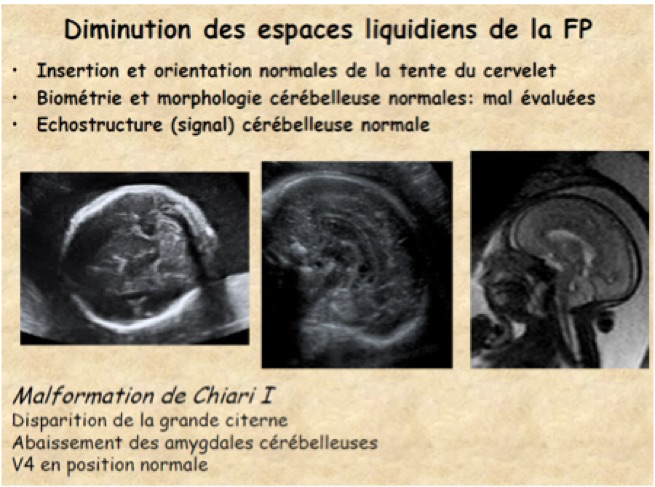
2-Rhombencéphalisynapsis
Il s’agit de la fusion des hémisphères et des pédoncules cérébelleux ainsi que des noyaux dentés associée à une agénésie vermienne.
En échographie, à l’étage sous-tentoriel on note:
-une petite fosse postérieure sans kyste ;
-un lobe cérébelleux unique hypoplasique, de taille variable et quelquefois asymétrique ;
-des sillons et des fissures orientés transversalement ;
-un vermis absent ou hypoplasique ;
-des flocculus présents mais hypoplasiques ;
-un V4 déformé en trou de serrure.
À l’étage sus-tentoriel on peut retrouver :
-une ventriculomégalie ;
-quelquefois une fusion des thalamus, du fornix ;
-en fait, tout peut se voir : agénésie calleuse, schizencéphalie, hétérotopies…
3-Dysraphie tectocérébelleuse
Exceptionnelle, sporadique, touchant plus volontiers le sexe masculin, elle consiste en une aplasie partielle ou totale du vermis cérébelleux et une traction dorsale des hémisphères cérébelleux et du tronc cérébral vers un defect osseux occipital.
En échographie, à l’étage sous-tentoriel on retrouve :
-une aplasie ou hypoplasie du vermis et des hémisphères cérébelleux ;
-une céphalocèle occipitale ;
-une désaxation postérieure du tronc cérébral, les hémisphères cérébelleux les contournant et se projetant en avant ;
-une déviation de la tente du cervelet et des sinus ;
-parfois une bifidité de l’écaille de l’occipital et de l’atlas.
À l’étage sus-tentoriel, on peut retrouver de façon variable : une agénésie du corps calleux, une hydrocéphalie, des troubles de la migration et de la giration.