I-Le spina biffida
A-Définition
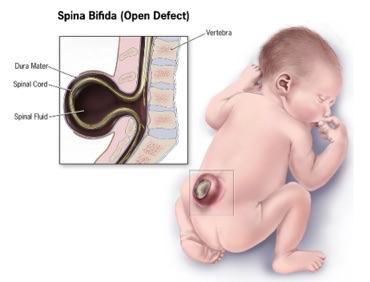
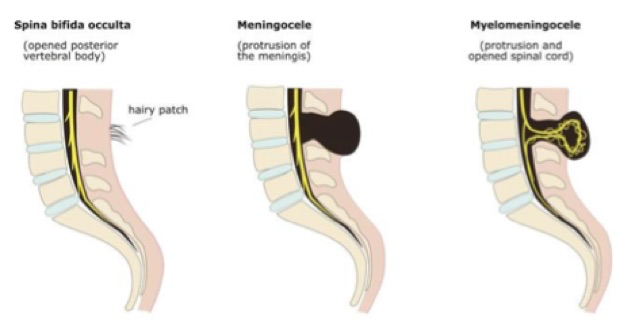
Le spina bifida (du latin signifiant « épine fendue en deux ») est une malformation liée à un défaut de fermeture du tube neural durant la quatrième semaine de la vie embryonnaire. Le plus souvent, cette malformation concerne l’extrémité caudalevoire lombaire. Il en résulte l’absence de l’apophyse épineuse d’une ou plusieurs vertèbres. La protrusion des méninges par cette ouverture donne un méningocèle. De gravité variable, ces malformations vont du spina bifida occulta au myéloméningocèle lorsqu’il y a protrusion de moelle épinière avec les méninges.
B-Signes échographiques
1-Signes directs
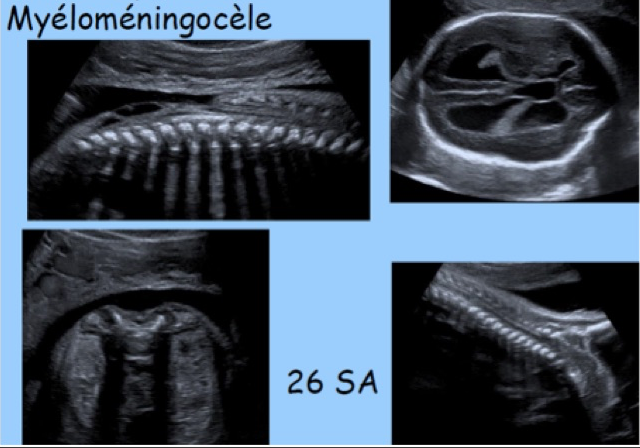
Dans plus de 90 % des cas, il s’agit de formes kystiques avec une composante liquidienne plus ou moins importante et saillante, correspondant le plus souvent à une myélo-méningocèle, plus rarement à une méningocèle. L’atteinte osseuse sera située le plus exactement possible en repérant sa hauteur ainsi que le nombre et le niveau des vertèbres atteintes. Par ordre de fréquence, l’atteinte est lombo-sacrée dans 80 % des cas, thoraco-lombaire dans 10 %, thoracique dans 7 % et cervicale dans 3 %. Au deuxième trimestre de la grossesse, les repères échographiques de la colonne fœtale sont les suivants : le corps vertébral de T12 est situé au niveau de la dernière côte et S1 correspond au sommet des ailes iliaques.

a-Coupe sagittale et parasagittale
Une solution de continuité du revêtement cutané et des parties molles est parfois évidente. Elle peut être dissimulée si le dos appuie sur la paroi utérine.
La hernie méningée, remplie de LCS, fait saillie en arrière du rachis et se repère souvent assez facilement mais on peut la manquer en fonction de la position du fœtus si elle est très petite ou, paradoxalement, très grande confondue alors avec une citerne de liquide amniotique.
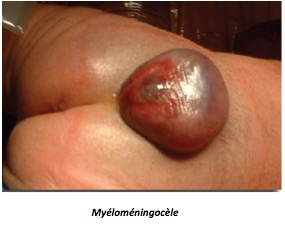
Le contenu est purement liquidien dans la méningocèle où la moelle n’est pas déplacée, mais, beaucoup plus souvent, il s’agit d’une myélo-méningocèle confirmée par la présence de travées nerveuses, échogènes, distribuées de façon linéaire ou anarchique.
La paroi est souvent fine, ce qui ne préjuge pas de sa nature histologique (méninge, plaque neurale, derme, épiderme), et elle est d’autant plus fine que la hernie est volumineuse avec une forte pression interne. Les épaississements, internes ou externes, et les gros plis évoquent clairement la présence d’une plaque neurale (myélo-méningocèle). En pratique, la présence et la qualité d’un revêtement cutané sont quasi impossibles à préciser.
Les lames latérales sont absentes ou irrégulièrement disposées sur une hauteur variable de la coupe parasagittale. C’est souvent cette irrégularité du « pointillé » rachidien qui attire en premier l’attention.
Une angulation rachidienne anormale est fréquente dans les larges spina, soit par déséquilibre de la statique vertébrale, soit par association avec une malformation des corps vertébraux.
b-Coupe transversale
-Ouverture des lames postérieures plus ou moins marquée, aspects classiquement décrits en U, en V ou en cupule (spina plan) selon le redressement ou la divergence des lames postérieures.
-Visibilité des cloisons dans la hernie méningée en cas de myélo-méningocèle.
c-Coupe frontale
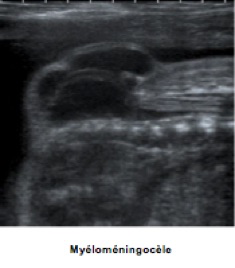
-Écartement des lames postérieures responsable de la perte de parallélisme du « rail spinal ».
-Affleurement et renflement anormal du canal rachidien qui perd son caractère linéaire et qui contient souvent une formation liquidienne allongée correspondant au « collet herniaire ».
-En dehors du rachis, tuméfaction (hernie) mobile avec le fœtus.
d-Formes particulières
Forme non kystique, spina plan ou myélocèle
Les signes osseux sont essentiels et le balayage transversal minutieux est parfois le plus efficace. Il n’y a pas de hernie liquidienne mais une plaque neurale plus ou moins nette au fond d’une cupule rachidienne. En coupe frontale, il n’y a pas de liquide entre les « rails » mais l’image échogène de la plaque neurale.
*Spina-lipome
Il est habituellement classé dans les spina-bifida occulta car le revêtement cutané est intact. C’est une masse lipomateuse sous-cutanée formant un amas échogène, homogène ou parfois kystique, occupant la ligne médiane et s’étendant en profondeur vers les vertèbres. Il est fréquemment associé à un spina-bifida occulta car il gêne la constitution de l’arc postérieur.
Parfois il s’associe à une hernie méningée réalisant une lipomyéloméningocèle et se traduisant par l’image d’une méningocèle revêtue d’un tissu épais et hyperéchogène. L’IRM fœtale constitue, dans ce cas, un examen complémentaire indispensable.
Le spina-lipome peut entraîner une anomalie de position du cône terminal qui, à l’état normal à la naissance, remonte approximativement au niveau de L1 (le canal rachidien s’allonge relativement beaucoup plus que la moelle). La moelle peut être fixée en position basse par la lésion ce qui va provoquer une myélopathie chronique appelée « syndrome de la moelle fixée ». En effet, lors des mouvements normaux du rachis, il existe une traction sur la moelle entraînant une compression du lit capillaire superficiel source de lésions anoxiques sous-jacentes irréversibles.
*Spina-bifida occulta (caché)
En l’absence de hernie et avec un defect osseux souvent minime, ces anomalies sont le plus souvent des découvertes post-natales et concernent plus les pédiatres que les obstétriciens. Le siège est souvent assez bas, volontiers sacré, ce qui explique les difficultés de visualisation prénatale. De plus, il n’y a habituellement pas d’anomalie cérébrale pour attirer l’attention de l’échographiste. Le diagnostic est souvent amené par l’existence d’une anomalie cutanée en regard du rachis.
*Myélocystocèle
Forme très rare, c’est une dilatation kystique du canal épendymaire, de mécanisme inconnu, qui entraîne une hernie myélo-méningée sous-cutanée, avec déhiscence de l’arc postérieur mais le revêtement cutanéo-adipeux est normal.
2-Signes indirects
-Anomalies de position des membres inférieurs
Elles sont la conséquence des déficits moteurs et des troubles du tonus éventuellement provoqués par la lésion médullaire. Elles sont donc inconstantes et ne constituent pas des signes d’appels intéressants. Il s’agit avant tout de malpositions des pieds et la mobilité des membres n’est pas obligatoirement perturbée.
-Anomalies du pôle céphalique
Ces anomalies sont la traduction échographique de la malformation type Chiari 2 . Il s’agit réellement de « marqueurs forts » de la dysraphie spinale, avec une forte valeur prédictive aussi bien positive que négative.
La biométrie céphalique (BIP et PC) est souvent diminuée et le BIP inférieur au 5e percentile témoigne d’un trouble associé de la croissance cérébrale. La grande citerne est inexistante, ce qui se traduit par l’impossibilité de visualiser la belle image des deux hémisphères cérébelleux L’effacement de la grande citerne peut (rarement) se rencontrer de façon isolée mais surtout, à l’inverse, la visibilité d’une grande citerne normale élimine un spina-bifida ouvert.
Les hémisphères cérébelleux sont attirés vers le bas et se glissent latéralement sur le côté du tronc cérébral, le cervelet s’enroule et se moule autour du tronc cérébral prenant un aspect en croissant à concavité antérieure, avec perte de relief des hémisphères, réalisant le classique signe de la banane.
D’autres modifications sont moins évidentes : le vermis inférieur ectopique (abaissé dans le trou occipital), le V4 aplati et étiré, l’horizontalisation de la tente du cervelet sur une coupe frontale postérieure.
Les deux os frontaux s’aplatissent ou prennent même parfois une forme biconcave, donnant un aspect plus ou moins pointu à la partie antérieure du crâne. C’est le « crâne citron ».
Une ventriculomégalie est très souvent associée. D’autres anomalies sont plus discrètes comme une agénésie partielle du corps calleux ou des troubles de la migration neuronale.
C-Pronostic et conduite à tenir
Tout va dépendre des réponses à ceratines questions.
1-Siège et étendue de la lésion
Nombre de vertèbres ouvertes. Un defect osseux correspondant à plus de trois arcs vertébraux est habituellement considéré comme péjoratif.
Hauteur de la première vertèbre ouverte Une lésion remontant au-dessus de L4 est un facteur défavorable. À l’opposé, une méningocèle sacrée a un pronostic plus favorable (mais risque de troubles sphinctériens).
2-Contenu de la lésion
Le diagnostic de myélo-méningocèle est porté par la présence au sein de la hernie méningée de travées échogènes. La méningocèle est totalement anéchogène. La myélo-méningocèle implique l’ouverture de la moelle sur l’extérieur avec des conséquences dégénératives pour le tissu nerveux exposé et des conséquences fonctionnelles importantes : le pronostic oscille entre médiocre et catastrophique (± paraplégie, ± incontinence, ± moelle fixée, ± hydrocéphalie, et aggravation avec la croissance).
3-Existe-t-il un retentissement ?
Une microcéphalie, ou simplement un céphalométrie au 3e-5e percentile, avec ventriculomégalie est de beaucoup plus mauvais pronostic qu’une ventriculomégalie isolée (atrophie cérébrale probable, troubles de la migration, etc.). De mauvais pronostic également : les malpositions des pieds, la mégavessie (difficulté de vidange vésicale), l’immobilité des membres inférieurs.
4-Existe-t-il des lésions ou pathologies extra-neurologiques associées ?
On en retrouve dans 30 % des cas et elles aggravent évidemment le pronostic :
-associations syndromiques avec cardiopathie, omphalocèle…
-anomalies chromosomiques : trisomie 13, 18, triploïdie. La dysraphie est alors rarement isolée dans ces cas. Devant toute anomalie de fermeture du tube neural, l’étude du caryotype fœtal est hautement souhaitable, pour aider le conseil génétique ultérieur et a fortiori si on envisage une poursuite de la grossesse. Le caryotype ne change rien, en général, à la prise en charge immédiate et une amniocentèse sera souvent réalisée juste avant l’interruption médicale de la grossesse. Accessoirement, le dosage l’alpha-fœtoprotéine amniotique et la caractérisation électrophorétique de l’acétylcholinestérase permettent d’affirmer le caractère ouvert ou fermé de la lésion, mais ces dosages sont moins pratiqués et ils n’interviennent plus dans le diagnostic des spina-bifida.
5-En pratique
L’interruption de grossesse est presque toujours proposée et acceptée. L’abstention est évidemment acceptée pour les raisons religieuses ou éthiques et il faut alors accompagner cette grossesse, proposer le caryotype, surveiller (une aggravation peut remettre en question la décision), préparer la naissance (rencontre avec neurochirurgien et associations). L’abstention peut également être proposée pour un spina-bifida occulta ou une méningocèle de petite taille, bas situé, sans signe cérébral associé, après concertation pluridisciplinaire.
II-Autres anomalies du rachis
A-Anomalies des vertèbres et de la courbure du rachis
On distingue les anomalies de segmentation au sens strict (bloc vertébral) et les anomalies de formation (hémi-vertèbres, vertèbres « papillon »), les deux pouvant être associées. Elles sont à l’origine de cyphoses et scolioses congénitales.
Sur le plan échographique, cela se traduit par un défaut d’alignement des centres d’ossification d’une ou de plusieurs vertèbres, en coupe sagittale et frontale.
Ces lésions sont, soit strictement isolées, soit associées à d’autres anomalies (cœur, reins, système digestif, extrémités) pouvant entrer dans le cadre d’associations diverses :
-syndrome VACTERL (1/6 000), association malformative hétérogène avec anomalie vertébrale (hémivertèbre, parfois spina), imperforation anale, cardiopathie, fistule trachéo-œsophagienne, anomalies radiales et rénales, amputation de segments de membres (limbs) ;
-syndrome d’Aicardi (exceptionnel 1/500 000), dominant lié à l’X, caractérisé par une agénésie du corps calleux constante et des anomalies de segmentation rachidienne, des anomalies rétiniennes, un retard mental et des convulsions ;
-malformation de Klippel Feil (1/35 000) : se caractérisant par une fusion C3-C4. Il peut exister une myélo-méningocèle cervicale, un pterigium colli, une cardiopathie, une surélévation de l’omoplate, une côte cervicale.
B-Diastématomyélie
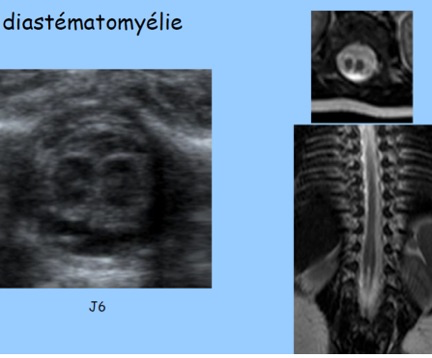
La diastématomyélie entre dans le cadre des dysraphismes fermés (spina-bifida occulta) et correspond à une division sagittale, non totale de la moelle en deux cordons, séparés ou non par un septum (éperon) ostéo-cartilagineux sur une hauteur variable. Le septum est en continuité avec la partie dorsale du corps vertébral et/ ou la partie ventrale de l’arc postérieur. Les deux cordons médullaires se rejoignent pour former le cône terminal en général unique. Le revêtement cutané est intact.
Il existe des associations fréquentes avec des anomalies du développement vertébral (diminution de leur diamètre antéro-postérieur, hémivertèbre, bloc vertébral) et des anomalies cutanées (télangiectasies, hémangiome, lipome, naevus, poils) sont parfois notées.
Les symptômes cliniques sont très variables et peuvent apparaître de façon tardive : syndrome neuro-orthopédique associant des troubles déficitaires et trophiques souvent asymétriques au niveau des membres inférieurs avec possibles pieds bots. Des problèmes sphinctériens existent en cas de moelle fixée basse. La réalisation d’une IRM fœtale et d’un scanner 3D complèteront le diagnostic échographique à la recherche de lésions médullaires ou osseuses. L’ablation de l’éperon en bas âge est conseillée pour éviter toute complication de fixation médullaire ultérieure.
En échographie, dans la forme isolée :
-voussure localisée sans anomalie du revêtement cutané ;
-angulation anormale du rachis en cas de vertèbres fusionnées ;
-lames latérales anormalement écartées en coupe frontale ;
-et, surtout, présence de l’éperon échogène séparant les cordons médullaires que l’on peut parfois deviner, formant un « troisième point d’ossification postérieur ».
C-Canaux et kystes neuroentériques
La persistance plus ou moins importante du canal chordal, aboutit à une fistule entre l’endoderme et l’ectoderme entraînant un développement anormal du tractus digestif qui reste en connexion avec les structures neurologiques médianes, souvent associé à un rachischizis antérieur. La fistule évolue le plus souvent vers la formation d’un (ou plusieurs) kystes d’une muqueuse de type digestif et qui sont toujours situés en avant de la moelle : pré-médullaires dans le canal rachidien, en avant du rachis et presque toujours dans le thorax.
Le diagnostic peut être logiquement évoqué devant une formation kystique intrarachidienne, volontiers associée à des malformations vertébrales (schizis antérieur, diastématomyélie). C’est beaucoup plus difficile quand le kyste a perdu le souvenir de ses origines et siège dans le thorax (souvent assimilé à un kyste bronchogénique ou une duplication digestive) ou l’abdomen, d’autant que l’anomalie vertébrale associée n’est pas obligatoirement en regard de la formation kystique.
D-Méningocèle antérieure
De localisation préférentiellement sacrée, elle suppose un rachischizis antérieur plus ou moins étendu ainsi qu’un defect méningé.
Elle forme une extension anéchogène antérieure, dans l’espace sous-péritonéal, d’une poche contenant du liquide cérébro-spinal : problème diagnostique d’un kyste pelvien.
Elle peut coexister – rarement – avec un tératome sacro-coccygien et peut alors évoquer un syndrome de Currarino (malformation obstructive ano-rectale, agénésie sacrée partielle, méningocèle et tératome pelvien), syndrome autosomique dominant à expressivité variable.
E-Rachischizis étendu et iniencéphalie
L’ouverture peut se situer à tous niveaux de la colonne vertébrale entraînant une angulation anormale du rachis. Du fait de l’origine au niveau du processus primordial de gastrulation, on conçoit la fréquence des associations avec d’autres anomalies malformatives. Le diagnostic est habituellement fait dès le premier trimestre et le caractère majeur de ces embryopathies rend recevable une proposition d’interruption de la grossesse.
1-Malformations « schizis type »
On classe ici les polymalformations associant diversement omphalocèle, anomalies diverses du tube neural, fente labiale et hernie diaphragmatique (anomalie de développement du mésoderme para-axial).
2-Iniencéphalie
Cette séquence malformative rare (0,1 à 10/10 000) se caractérise par une hypoplasie ou une absence de l’écaille occipitale, un élargissement du trou occipital, un dysraphisme cervical (rachischizis des premières vertèbres cervicales), une déflexion extrême de la tête avec lordose cervico-thoracique et une déformation du rachis cervical par fusion ou agénésie de plusieurs corps vertébraux. Dans l’iniencéphalie « ouverte », il existe une encéphalocèle postérieure. Dans l’iniencéphalie « fermée », le cuir chevelu occipital est intact mais se poursuit directement avec la peau dorsale.
Sur le plan échographique, le cou n’est pas identifié et la tête semble en continuité directe avec le tronc, face et yeux au zénith. Les anomalies associées sont multiples :
-surtout cérébrales : hydrocéphalie, microcéphalie, holoprosencéphalie, agénésie vermienne, kyste cérébelleux ;
-mais aussi anomalies costales, célosomie, hernie diaphragmatique, fente labio-palatine, malformations cardiaques.
3-Limb body wall complex
Syndrome polymalformatif complexe, et sans symétrie, que l’on assimile souvent à une forme majeure de maladie des brides amniotiques.
Les adhérences au niveau du névraxe sont à l’origine de defects majeurs : exencéphalie, rachischizis et/ou myélo-méningocèle étendus, scolioses ou cyphoses importantes, fentes faciales non systématisées.
Il s’y associe diversement:
-des anomalies pariétales souvent majeures (célosomies abdominales et/ou thoraciques) ;
-des anomalies anarchiques des membres (amputations, déformations à tous les niveaux) ;
-des anomalies uro-génitales (imperforation anale) ;
-des brides (inconstantes).
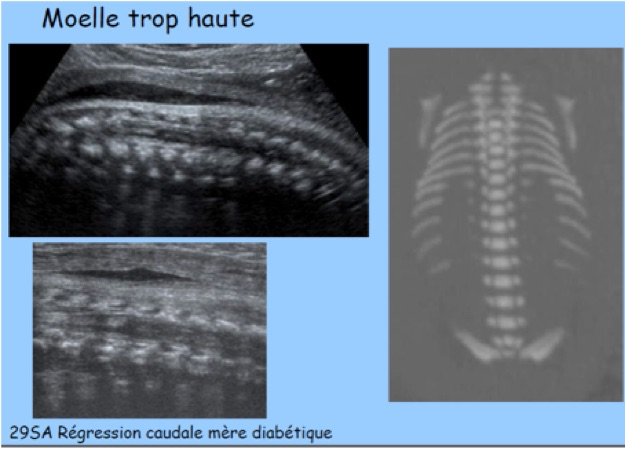
F-Syndrome de régression caudale
Le terme de « syndrome de régression caudale » regroupe un ensemble de malformations congénitales rares allant de la simple agénésie sacrée à la fusion complète des deux membres inférieurs, forme la plus sévère. Il est souvent évoqué un rôle du diabète maternel.
Le bourgeon caudal se transforme en appendice caudal, formation transitoire qui atteint son maximum de développement vers 7,5 SA avant de régresser complètement avant 9 SA. Le mésenchyme du bourgeon est à l’origine de la partie terminale de l’axe vertébro-médullaire (neurulation secondaire) mais aussi du plancher périnéal antérieur et postérieur. La régression de l’appendice caudale est un phénomène normal et nécessaire, le terme de « syndrome de régression » est donc un peu ambigu : il désigne en réalité soit la disparition trop précoce de l’appendice (exagération du processus normal), soit son absence de développement (avortement du bourgeon caudal). La conséquence de l’absence de bourgeon caudal est le rapprochement et la fusion des ébauches des membres inférieurs qui constitue la manifestation la plus spectaculaire de ce syndrome : la sirénomélie. Dans d’autres formes moins sévères, il existe une agénésie sacrée avec abduction importante des cuisses (attitude en Bouddha) qui constituerait pour certains auteurs le véritable syndrome de régression caudale classique, de mécanisme différent de la sirénomélie.
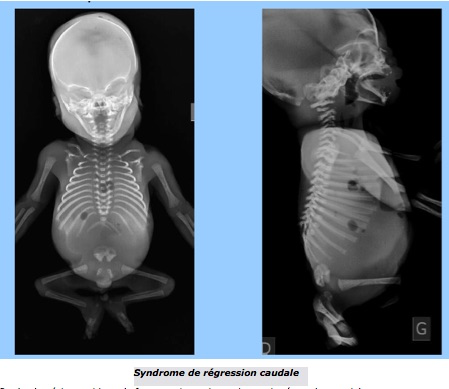
Sur le plan échographique, la forme majeure de syndrome de régression caudale, avec sirénomélie, s’accompagne d’une agénésie rénale bilatérale quasi constante, responsable d’un oligo-anamnios avec séquence de Potter : insuffisance rénale et oligo-anurie → oligoamnios → compression fœtale → faciès aplati, peau fripée et, surtout, hypoplasie pulmonaire (mais aussi pieds bots et luxation de hanche dans les autres étiologies d’oligo-anurie).
Le reste des signes échographiques découle de la séquence malformative et associe de façon variable :
-une atteinte vertébrale : interruption brutale de l’image du rachis (absence simple de sacrum, agénésie lombo-sacrée parfois étendue aux vertèbres dorsales), hémi-vertèbres ; méningocèle ou myélo-méningocèle ;
-une atteinte digestive : agénésie rectale, imperforation anale ;
-une atteinte du tubercule génital : agénésie, aplasie vulvaire, états asexués ;
-une atteinte des canaux de Wolf et de Muller : absence de bourgeon urétéral, agénésie rénale uni ou bilatérale, agénésie vaginale ou utérine ;
-une atteinte de l’allantoïde : agénésie vésicale, artère ombilicale unique naissant directement de l’aorte ;
-une atteinte des ébauches des membres inférieurs : absence des os longs à l’exception d’un fémur, abduction et rotation externe (péronés – s’ils existent – en dedans du tibia), luxation de hanche, pieds bots, atrophie.
Le syndrome OEIS associe des éléments de régression caudale et un spina-bifida
Le syndrome de Townes-Brocks est une forme génétique de syndrome de régression caudale avec un gène responsable identifié sur le chromosome 16. Il associe :
-une anomalie de type régression caudale (imperforation anale, malformations des pieds, anomalies vertébrales) ;
-des malformations de l’oreille externe (appendices prétragiens, hélix malformés, sténose du conduit auditif externe) ;
-une anomalie de l’axe radial (pouce triphalangé, pouce bifide, hexadactylie) ;
-des anomalies rénales et cardiaques.
G-Tératome sacro-coccygien
Le tératome sacro-coccygien est une tumeur se développant à partir de cellules embryonnaires pluripotentes de la région caudale. Sur le plan histologique, on distingue les tératomes bénins (matures, différenciés) et les tératomes malins (immatures). La plupart des tumeurs découvertes en période anténatale sont bénignes. La lésion est relativement rare : un cas pour 35 000 à 40 000 naissances, touchant quatre fois plus les filles que les garçons.
1-Aspect échographique et malformations associées
Dans sa forme isolée typique, il se présente sous l’aspect d’une tumeur solide ou mixte, plus rarement kystique (15 % des cas), souvent volumineuse et dont l’implantation se situe au niveau sacré ou sacro-coccygien. Le tératome est volontiers volumineux d’emblée (aussi gros que la tête fœtale à 22 semaines), ses contours sont irréguliers. Des calcifications sont parfois associées . Le rachis est strictement normal, ce qui permet de le différencier de la méningocèle et de la myélo-méningocèle. Le pôle céphalique est également normal.
Le Doppler met en évidence une vascularisation importante à partir d’une large artère sacrée (parfois aussi large que l’aorte) dont on retrouve le trajet initial en position médiane en avant du sacrum.
La classification d’Altman est la plus couramment utilisée et elle regroupe les tératomes en quatre types :
-type I : tératome extra-pelvien avec composante pré-sacrée minime ;
-type II : tératome extra-pelvien avec composante intra-pelvienne nette (Figure 10-20.f) ;
-type III : tératome extra et intra-pelvien avec extension abdominale ;
-type IV : tératome intra-pelvien.
Les anomalies associées (près de 20 % des cas) touchent le système musculo-squelettique et surtout le rachis inférieur : hypoplasie du sacrum, spina-bifida occulta, hypoplasie de L5, spondylolyse, spondylolisthésis, blocs vertébraux ou fusion partielle L4-L5. Les formes à composante intra-pelvienne sont responsables de phénomènes de compression au niveau des organes de voisinage (hydronéphrose, voire dysplasie rénale, dilatation digestive). Actuellement, l’IRM fœtale s’impose pour préciser les connexions et l’extension de la lésion.
Le tératome sacro-coccygien de type IV peut poser des problèmes de diagnostic différentiel avec un kyste de l’ovaire, un pseudo-kyste méconial, une duplication digestive, une méningocèle sacrée antérieure.
2-Complications
Les principales complications sont l’hydramnios et l’anasarque fœto-placentaire par décompensation cardiaque. Un hydramnios aigu peut être une circonstance de découverte. Pour expliquer leur survenue, Nyberg retient trois mécanismes : l’existence d’un shunt artério-veineux au sein de la tumeur, la constitution d’une anémie fœtale par hémorragie intra-tumorale ou encore la transsudation de liquide à partir du tératome. Le risque semble d’autant plus élevé que le degré de vascularisation de la tumeur est important.
L’hémorragie intra-tumorale se traduit par l’apparition de plages liquidiennes au sein de la tumeur.
La mort fœtale in utero est possible, en rapport avec une décompensation cardiaque ou une rupture vasculaire.
3-Critères pronostiques
Ce sont les suivants : taille de la tumeur,Struture solide, importance de la vascularisation, apparition d’un hydramnios ou d’une anasarque fœto-placentaire est très péjorative, croissance rapide de la tumeur.