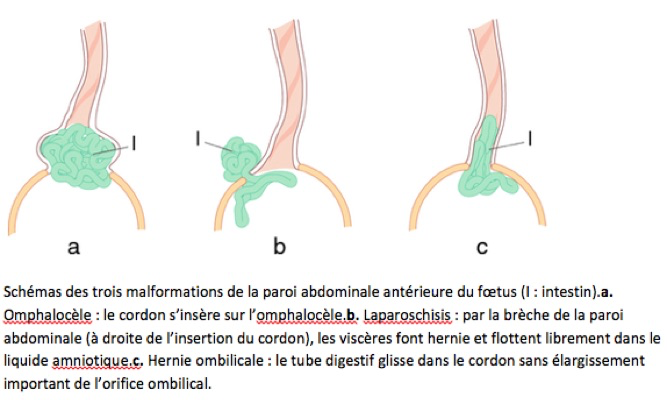
L’absence de fermeture de la paroi abdominale antérieure est appelée cœlosomie. Selon l’origine du défaut defermeture, on distingue:
–les cœlosomies moyennes représentée typiquement par l’omphalocèle et le laparoschisis;
-les cœlosomies supérieures qui associent le plus souvent une omphalocèle avec une ouverture du sternum et du diaphragme entraînant une ectopie cardiaque ;
–les cœlosomies inférieures qui peuvent donner une exstrophie vésicale isolée ou, dans des formes plus complètes, être associée une omphalocèle sous-ombilicale et/ou un défaut de cloisonnement du cloaque (réalisant une exstrophie cloacale plutôt que vésicale) ;
Diverses combinaisons sont possibles entre les trois formes, jusqu’à la célosomie totale.
Nous n’envisageons ici que les principales.
Laparoschisis
Définition
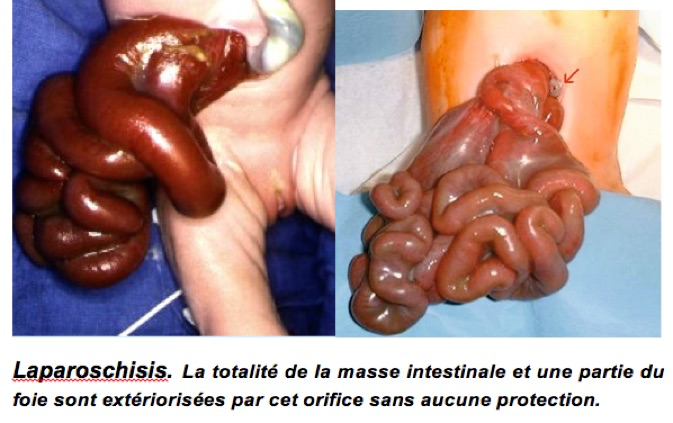
Le laparoschisis, aussi appelé « gastroschisis », est une malformation consistant en une fente de la paroi abdominale à travers laquelle passent les intestins. Il n’existe aucune membrane entourant les intestins. Ces derniers flottent dans le liquide amniotique, sans protection. L’anomalie se forme vers 10-12 semaines de grossesse par un arrêt de croissance localisé de la paroi abdominale (aplasie, nécrose). Elle se situe très souvent sur le côté droit de l’ombilic. La taille de la fente excède rarement 30 millimètres.
Fréquence
Sa fréquence varie selon les séries de 1 pour 3 000 à 1 pour 10 000 naissances vivantes.
Causes
Aucune anomalie chromosomique n’a été mise en évidence comme cause du laparoschisis. Le laparoschisis peut apparaître à la suite d’un traitement par pseudoéphédrine (vasoconstricteur) par voie orale. Le jeune âge de la mère est également un facteur de risque. On observe d’autre part que l’anomalie est particulièrement plus fréquente dans les ïles Hawaï à proximité de zones où des multinationales phytosanitaires comme Syngenta, Bayer, Monsanto, Dow, BASF ou Dupont réalisent des tests d’OGM ou d’épandages de pesticides.
Le laparoschisis est le plus souvent isolé. Des malformations associées extra-digestives sont retrouvées dans 6 % des cas. Les malformations digestives (à type d’atrésies ou de sténoses, de grêle court, de persistance du diverticule de Meckel…) sont un peu plus fréquentes (15 %). Les anomalies chromosomiques en revanche sont exceptionnelles rendant inutile la réalisation d’un caryotype. Le phénomène est essentiellement local. Le pronostic est lié à la qualité et à la vitalité des anses digestives éviscérées.
Le diagnostic anténatal
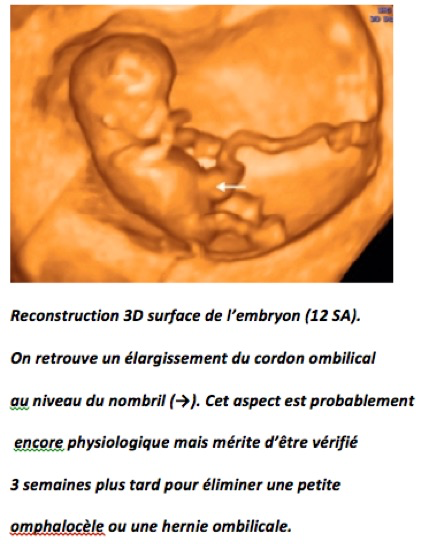
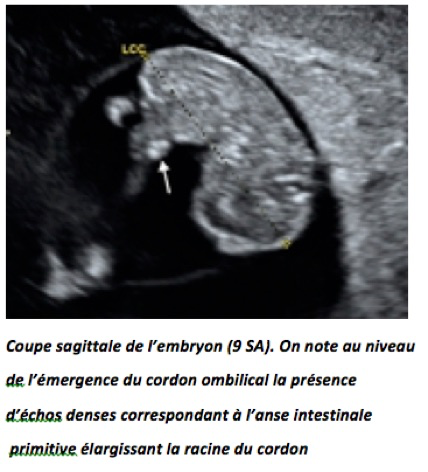
La paroi abdominale ne se fermant complètement qu’à 12 semaines, le diagnostic de laparoschisis ne peut être fait avant cette période. Le développement de l’anse iléale primitive est caractérisé par son allongement extrêmement rapide qui l’oblige à se développer en dehors de l’abdomen en faisant hernie dans le cœlome extra-embryonnaire du cordon à travers l’ombilic, c’est la classique hernie ombilicale physiologique qui est visible en échographie de la 8ème à la 12ème semaine. L’élargissement de la cavité abdominale associée à une diminution de la taille relative du foie et des reins permet une réintégration des anses intestinales : c’est le phénomène de réduction. Après 12semaines, en échographie, on ne doit plus mettre en évidence de hernie ombilicale.
L’échographie du deuxième trimestre montre:
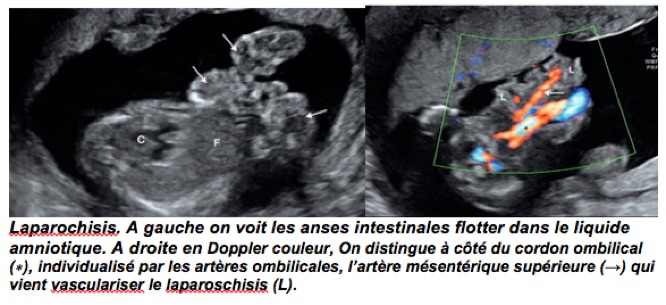
-la présence de plusieurs anses intestinales, herniées à travers un orifice para-ombilical droit, dont il faut mesurer le diamètre, et flottant librement dans le liquide amniotique, sans membrane recouvrant la malformation ;
-le cordon ombilical est normalement inséré à gauche du defect pariétal et son trajet est confirmé en Doppler couleur
-plus rarement, d’autres organes peuvent se hernier comme l’estomac, le foie, la vésicule biliaire, la vessie. Toutefois, la présence d’une éviscération de ces organes doit faire craindre une malformation plus complexe (syndrome du cordon court, maladie des brides amniotiques, exstrophie cloacale) et doit faire rechercher d’autres malformations associées.
Eléments de pronostic
La vitalité des anses digestives est le principal élément du pronostic. Les lésions intestinales sont dues à l’irritation produite par le liquide amniotique entraînant un épaississement des anses intestinales herniées (péritonite chimique) qui finissent par s’agglutiner de manière compacte. La striction progressive au niveau du collet (l’intestin grandit plus rapidement que le defect pariétal) peut être responsable d’une anomalie du retour veineux et lymphatique avec œdème, ou d’une ischémie pouvant conduire à une sténose, une atrésie, une nécrose et une perforation intestinale. Ces lésions se développent surtout en fin de grossesse.
Bien que ne faisant pas l’objet d’un consensus dans la littérature, on tentera d’apprécier l’état de l’intestin sur:
-Le diamètre des anses herniées : la lumière intestinale du grêle est considérée comme dilatée au-delà de 6 mm, ce qui est souvent prédictif de complications post-natales. Au-delà de 17 mm, la morbidité est élevée avec notamment un risque d’atrésie du grêle.
-Mais l’état de l’intestin intra-abdominal est également important : une dilatation « asymétrique » des anses grêles intra-abdominales sans dilatation des anses éviscérées est également de mauvais pronostic.
-L’apparition d’un hydramnios.
-L’épaississement et l’échogénicité de la paroi intestinale : au-delà de 3 mm, il s’accompagne d’une plus grande morbidité post-natale.
-La taille de l’orifice pariétal : une ouverture pariétale étroite (inférieure à 10 mm) augmente le risque d’ischémie intestinale par strangulation des anses et du mésentère.
-La vascularisation des anses herniées : il est possible de l’apprécier en recherchant un signal Doppler au niveau de la paroi digestive, notamment par Doppler énergie, et en mesurant l’index de résistance artérielle ou de pulsatilité de l’artère mésentérique supérieure dans sa portion herniée et dans sa portion intra-abdominale au niveau de son origine et de l’ouverture pariétale.
Evolution
La surveillance échographique du laparoschisis doit être mensuelle puis hebdomadaire dans les deux derniers mois. Elle permet ainsi de suspecter une complication :
-oligoamnios (par souffrance fœtale charonique) ou à l’inverse hydramnios (par occlusion digestive haute) ;
-retard de croissance fœtale (méfiance dans l’estimation pondérale faussée par un abdomen déshabité) ;
-si l’aspect des anses intestinales extériorisées se modifie en s’agglutinant devant l’orifice pariétal alors qu’elles étaient auparavant libres dans le liquide amniotique avec péristaltisme et lumière digestive visible;
-extériorisation de la vessie par l’orifice du laparoschisis augmentant le risque de compression intestinale.
Traitement
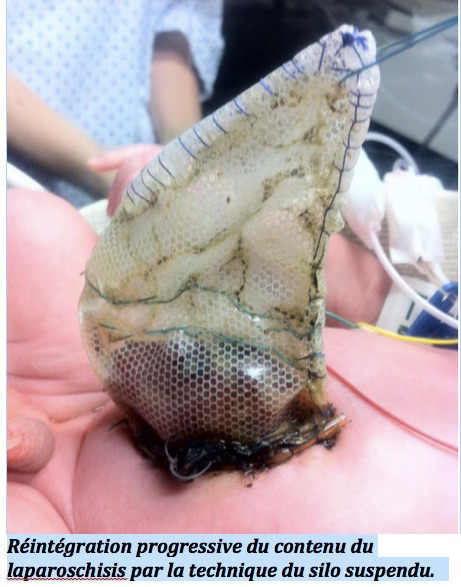
L’accouchement est fait par césarienne dans un centre spécialisé, ce qui facilite très largement la prise en charge. La réintégration des anses intestinales se fait immédiatement ou progressivement. La réintégration des anses se fait si possible en un temps ou sinon de façon séquentielle avec la confection d’un silo avec des plaques de Silastic suturées à la peau du collet avec un fil non résorbable et réunies en haut au-dessus des viscères herniés et entourées de compresses stériles. Le silo est suspendu à la couveuse pour que la gravité fasse rentrer les anses. La fermeture de l’appareil est fait toutes les 24 heures, progressivement, sous strictes conditions d’asepsie.La fermeture définitive est faite au bloc opératoire au bout d’ube semaine environ. En règle générale, une réalimentation complète par la bouche est obtenue au bout de six semaines. En attendant, la nutrition se fait par voie veineuse centrale.
L’omphalocèle
Définition
Il s’agit d’un défaut de fermeture ventrale ou célosomie moyenne, concernant l’orifice ombilical lui-même donc de siège central. L’aplasie plus ou moins large de la paroi intéresse tous les plans : péritoine, muscles et peau. Le contenu de l’abdomen, intestin et foie notamment, hernié par cet orifice lié à l’aplasie forme une tuméfaction au sommet de laquelle s’implante le cordon. Cette tuméfaction est recouverte et protégée par une fine membrane anhiste qui n’est autre que la base du cordon élargie par cette hernie. La masse intestinale est par ailleurs normale.
Fréquence
Sa fréquence est environ de 1/4 000 grossesses et elle augmente avec l’âge maternel.
Signes échographiques
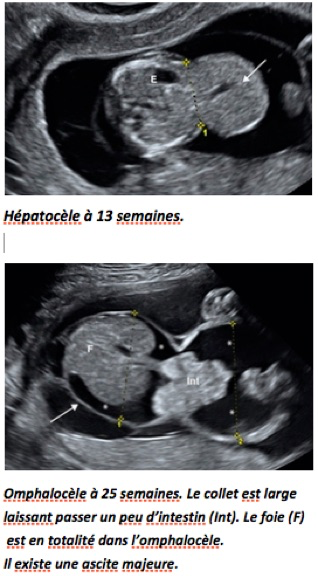
-un élargissement de l’orifice ombilical, avec la présence d’une masse bien limitée, appendue à la paroi abdominale antérieure et cernée d’une fine membrane;
-l’insertion du cordon sur la membrane;
-le contenu est de l’intestin grêle échogène et souvent une partie du foie un peu moins échogène voire tout le foie. La présence de la veine ombilicale visible dans le sac signant la présence de foie (on parle d’hépatocèle).
Anomalies associées
Le diagnostic d’omphalocèle étant posé, il faut rechercher les associations malformatives et anomalies chromosomiques.
a-Anomalies chromosomiques
La réalisation d’un caryotype doit être systématique, du fait de la fréquence (environ 30 %) des anomalies chromosomiques associées (trisomie 18 et 13, plus rarement 21, triploïdie et monosomie X).
b-Malformations associées
-malformations cardiaques (20 % à 50 %) surtout dans les omphalocèles volumineuses et supérieures, rarement dans les célosomies inférieures: tétralogie de Fallot, communication interventriculaire, communication interauriculaire, canal atrioventriculaire, cardiopathies complexes ;
-malformations génito-urinaires (17 %), surtout en cas d’association avec une célosomie inférieure (exstrophie vésicale, voir infra) : dysplasies rénales, méga-uretère, reins polykystiques ;
-malformations du système nerveux central (7 %) et de la face (16 %) : anencéphalie, microcéphalie, fentes labio-palatines, myélo-méningocèle ;
-malformations des membres et des extrémités (6 %): polydactylie, syndactylie, pied-bot, amélie ;
-malformations intestinales paradoxalement beaucoup plus rares (1 %) : atrésies intestinales, Meckel.
c-Associations syndromiques
Ces malformations viscérales peuvent se regrouper en syndromes plus ou moins complexes.
–Le syndrome de Beckwith-Wiedemann, dont la fréquence est estimée à 1/14 000 naissances, se caractérise par une croissance excessive du fœtus et associe de façon variable : omphalocèle (75 % des cas), macroglossie, macrosomie avec viscéromégalie, hépatosplénomégalie (32 %) ou néphromégalie (23 %), cardiopathies (15 %). De nombreuses autres anomalies sont plus rarement retrouvées : microcéphalie, dysmorphie faciale, exophtalmie, occiput proéminent, hémi-hypertrophie totale ou partielle, hernie diaphragmatique, malformation génito-urinaire, tumeur hépatique, rénale (tumeur de Wilms) ou surrénalienne, hémangiome ou placenta kystique. La complication néonatale essentielle est l’hypoglycémie secondaire à un hyperinsulinisme. La mortalité est de 20 %, secondaire à cette hypoglycémie mais aussi aux complications des malformations associées. Le gène est identifié : ce syndrome est secondaire à une anomalie sur le locus p15.5 du chromosome 11. La majorité des cas sont sporadiques (85 %), les autres cas sont des formes familiales.
–La pentalogie de Cantrell entre dans le cadre des célosomies supérieures.
Ce syndrome polymalformatif exceptionnel, dans la plupart des cas sporadique, dépisté dès le 1er trimestre de grossesse, associe dans sa forme complète :
– une omphalocèle sus-ombilicale (pouvant contenir l’estomac),
– une ectopie cardiaque partielle (le cœur est engagé dans l’omphalocèle),
– une malformation sternale inférieure (agénésie ou fente),
– une ouverture diaphragmatique (entraînant une hernie diaphragmatique si elle est importante),
– une ouverture du péricarde apical,
– l’association est possible avec d’autres malformations (fente labiale ou labiopalatine, encéphalocèle, exencéphalie, sirénomélie, cardiopathie notamment tétralogie de Fallot). Une aberration chromosomique peut être associée. Le pronostic est évidemment désastreux.
Ce syndrome est à différencier de :
– l’ectopie cardiaque (éviscération d’un cœur généralement malformé au travers d’une ouverture pariétale, sternale et/ou péricardique), souvent associée à une omphalocèle et à des anomalies craniofaciales et chromosomique type trisomie 18,
– l’agénésie sternale isolée : la paroi antérieure du thorax est pulsatile, synchrone des battements cardiaques. Le cœur peut paraître trop antérieur mais la paroi du thorax est intacte. Il peut s’y associer un hémangiome pariétal masquant le diagnostic.
–Le syndrome OEIS, exceptionnel (1/200 000 naissances), associe une Omphalocèle, une Exstrophie vésicale, une Imperforation anale et un Spina bifida (large myéloméningocèle lombo-sacrée) visualisé directement ou suspecté sur les signes intracrâniens (malformation de Chiari). Il peut s’y associer une scoliose, des pieds bots et un thorax étroit.
–Le syndrome du cordon court. D’une fréquence estimée à 1/30 000 grossesses, il est probablement plus fréquent car il est responsable d’avortements spontanés faits avant le diagnostic échographique. Le cordon ombilical paraît court voire absent (recherché en Doppler couleur). Les viscères abdominaux sont extériorisés dans l’espace extracœlomique (cœlome externe) et sont accolés plus ou moins étroitement à la surface placentaire. Ce syndrome associe de larges defects crâniens, une malformation de la paroi thoracique et/ou abdominale, des amputations des membres, une cyphoscoliose, des malformations viscérales et un oligoamnios. Il n’est généralement pas retrouvé d’anomalie chromosomique. On en rapproche le limb body wall complex (ou syndrome) : séquence cordon court (abdominoschisis, scoliose) ± exencéphalie, fente labiale, ectromélie, imperforation anale ± anomalies génitales ± myéloméningocèle. On peut aussi en rapprocher la grossesse extra-membraneuse et le syndrome des brides amniotiques qui associe, dans sa forme complexe, asymétrie faciale avec exencéphalie, amputations des doigts et des orteils, abdominochisis.
Conduite à tenir
Pour la naissance, la césarienne est conseillée si le diamètre de l’omphalocèle est supérieur à 7cm. En dessous, la voie basse peut être envisageable. Le conditionnement dès la naissance est bien sur capital. Le pronostic va dépendre du volume et des anomalies associées. La réintégration peut faire appel:
-à des procédés naturels de réépidermisation (c’est le tannage);
-à la méthode de Schuster qui utilise un silo de silastic;
-à une fermeture chirurgicale simple ou avec plaque;
-à la technique de Yazbeck qui utilise des champs collés resserrés progressivement.
Le pronostic reste mauvais pour les omphlocèles géantes du fait notamment des complications respiratoires.
La hernie ombilicale
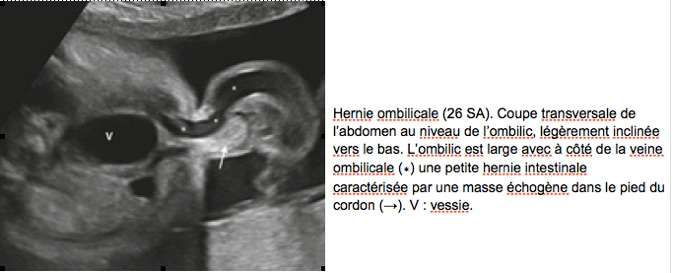
La hernie ombilicale correspond à un défaut de réintégration sans élargissement important de l’orifice. La hernie n’est pas toujours visible in utero mais on la craint toujours chez le nouveau-né – même si le phénomène est rare –, ce qui conduit à toujours clamper le cordon à quelques centimètres de son insertion par crainte de pincer une anse intestinale.
Lorsque le contenu est purement intestinal (30 % des cas) et de petit volume, on peut discuter le diagnostic de hernie ombilicale où dans un ombilic large s’extériorise au minimum une anse intestinale très échogène, mais la peau et l’insertion du cordon sont normales. Les examens complémentaires à proposer seront les mêmes que pour une omphalocèle intestinale.
L’exstrophie vésicale
Elle fait partie des célosomies inférieures et se traduit par un defect pariétal bas avec une ouverture de la vessie : la muqueuse vésicale, plus ou moins végétante, fait saillie à ce niveau et se poursuit directement avec la peau avoisinante. Cette malformation, retrouvée dans 1/30 000 naissances, est plus fréquente chez le garçon, avec des conséquences également plus graves sur le plan urinaire et sexuel.
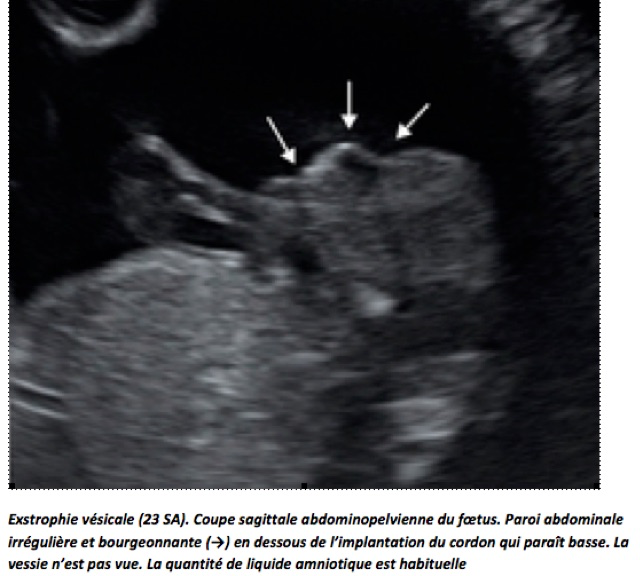
En échographie, le signe essentiel est l’absence permanente de visualisation de la vessie, alors que le haut appareil urinaire est normal ainsi que la quantité de liquide amniotique. La muqueuse vésicale saillante boursoufle de façon irrégulière la région sous-ombilicale qui prend un aspect « fripé ». L’examen Doppler couleur peut aider à localiser la plaque vésicale en montrant les artères ombilicales dans le pelvis.