Le retard pubertaire chez la fille se définit par l’absence de développement des seins après l’âge de 13 ans et/ou l’absence de survenue des menstruations après l’âge de 16 ans ou 4 ans après le début du développement mammaire.
Il existe donc deux situations : l’impubérisme (absence de développement des seins) et l’aménorrhée primaire (développement des seins présents, mais pas de règles).
Nous traitons dans ce chapitre de l’impubérisme.
Conduite à tenir
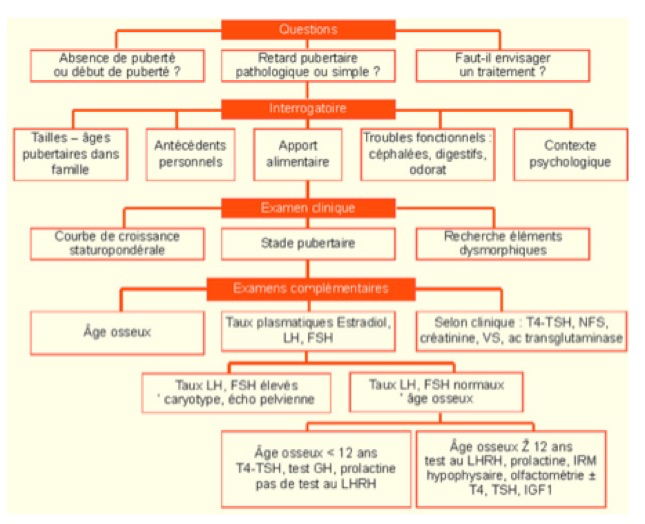
Devant un impubérisme, il faut rechercher à l’interrogatoire des antécédents familiaux de retard pubertaire, un trouble de l’olfaction, des signes digestifs, des troubles du comportement alimentaire, à l’examen clinique : un retard staturo-pondéral, des signes généraux (pâleur, adénopathies, etc.) des signes dysmorphiques en faveur d’un syndrome de Turner, des céphalées, des troubles visuels évocateurs d’une hypertension intracrânienne.
Les premiers examens à réaliser sont :
-un âge osseux,
-un dosage de FSH et de LH qui permettra de déterminer si l’hypogonadisme est d’origine périphérique ou centrale. Si le taux de FSH et de LH est élevé ; il s’agit d’une insuffisance ovarienne. Le caryotype s’impose car la première cause à chercher est le syndrome de Turner (surtout si un retard de croissance est associé). Si le caryotype est normal, il faut rechercher une auto-immunité par la recherche d’atteinte auto-immune associée; le dosage des anticorps anti-ovariens n’est pas spécifique. Il faut également réaliser une échographie pelvienne pour évaluer la taille des ovaires. Si le bilan auto-immun est négatif, une recherche de cause génétique plus rare est à proposer.
Si le taux de LH et FSH est normal, il faut alors évaluer l’âge osseux.
La radiographie de la main permet de définir un âge osseux si les caractères sexuels secondaires sont peu développés ou absents (le sésamoïde (cf. glossaire) du pouce apparaît pour un âge osseux de 13 ans).
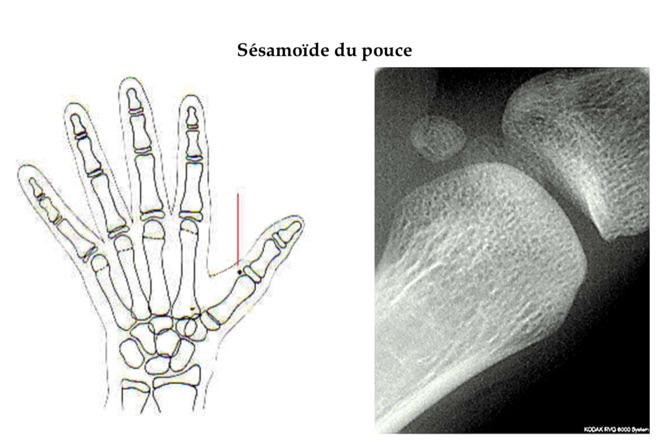
-Si l’âge osseux est ≥ 12 ans, il faut rechercher un déficit gonadotrope par un test au LHRH et éventuellement d’autres déficits hypophysaires associés (GH, TSH et ACTH). Si le déficit gonadotrope est confirmé, une IRM cérébrale doit être réalisée à la recherche d’une tumeur ou d’une anomalie des bulbes olfactifs (en faveur d’un syndrome de Kallmann).
-Si l’âge osseux est < 12 ans, le déficit gonadotrope est difficile à diagnostiquer car le test au LHRH est difficilement interprétable et n’a donc pas lieu d’être réalisé. Souvent, ce tableau est associé à un retard statural nécessitant la recherche d’un déficit en hormone de croissance (GH) ou d’une hypothyroïdie. Si ce bilan est normal, le diagnostic le plus probable est un retard pubertaire simple qui sera confirmé par l’évolution clinique.
La cause des impubérismes peut donc être centrale ou ovarienne. Le dosage des gonadotrophines FSH et LH de base ou le test au LHRH permet de confirmer son origine centrale ou périphérique.
Les causes de l’impubérisme
A-Les insuffisances gonadiques
C’est l’insuffisance du fonctionnement des ovaires qui est en cause. Ces insuffisances peuvent être congénitales ou acquises. Les insuffisances gonadiques sont responsables d’environ 40 % des retards pubertaires chez la fille. Elles se caractérisent par un taux de FSH et de LH élevé.
1-Insuffisances gonadiques congénitales
a-Le syndrome de Turner. C’est la cause la plus fréquente. Il affecte 1/2 500 nouveaunés de sexe féminin. Il faut y penser devant un retard de croissance intra-utérin, une petite taille (taille inférieure à -2 déviations standard et/ou un ralentissement de la vitesse de croissance) et des signes dysmorphiques qui peuvent être discrets. Le caryotype est 45 XO ou en mosaïque, ou encore comporte une anomalie de structure d’un chromosome X. L’insuffisance ovarienne affecte plus de 95 % des patientes. Un démarrage pubertaire spontané peut s’observer chez jusqu’à 30 % des patientes (surtout les formes avec mosaïcisme), avec des menstruations spontanées dans 2 à 5 % des cas. Dans la majorité des cas, cette activité ovarienne cesse rapidement : absence de progression de la puberté, aménorrhée secondaire. Des grossesses spontanées ont été exceptionnellement décrites.
Le diagnostic peut être évoqué à divers stades :
-en anténatal, les signes d’appel échographique sont : nuque épaisse, hygroma kystique, hydrops fetalis (anasarque), anomalies cardiaques gauches (Bicuspidie aortique, coarctation aortique , sténose aortique,insuffisance aortique, prolapsus mitral , hypoplasie du cœur gauche, dilatation aortique (± anévrisme, ± rupture), anomalies rénales, Brachycéphalie Polyhydramnios, oligohydramnios, retard de croissance modéré.
-chez le nouveau-né de sexe féminin : lymphœdème des extrémités ;
-chez nourrisson, enfant de sexe féminin : déficit statural (taille ≤ -2 DS ou taille ≤ -2 DS par rapport aux tailles moyennes parentales) quelle que soit la vitesse de croissance, ou ralentissement statural, avec ou sans phénotype clinique évocateur de ST (annexe 1), antécédent de coarctation aortique ;
-chez adolescente : déficit statural ≤ -2 DS avec ou sans phénotype clinique évocateur (annexe 1), retard pubertaire avec absence de développement mammaire après l’âge de 13 ans, non progression du développement pubertaire, aménorrhée primaire ou secondaire avec élévation des gonadotrophines sériques ;
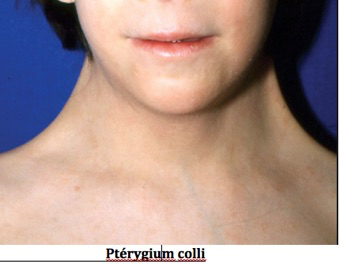
-Chez l’adulte : petite taille, phénotype clinique évocateur (dysmorphie cranio-faciale : visage triangulaire, hypoplasie du maxillaire inférieur, rétrognatie, oreilles basses), cheveux implantés bas, un cou court large et bridé quasi « palmé » (c’est le classique pterygium colli), un thorax large « en bouclier » (pectus excavatum), un écartement mamelonnaire.
Le profil hormonal est caractérisé par une élévation franche de la FSH et de la LH et des taux bas d’estradiol .
Le diagnostic de certitude est établi après la réalisation d’un caryotype qui retrouve une monosomie 45,X dans environ 50 % des cas, les autres formes étant constituées essentiellement par des formes en mosaïque (45,X/46,XX, etc.) et plus rarement par des anomalies de structure du chromosome X.
La prise en charge est multidisciplinaire et consiste d’abord à l’évaluation des différentes anomalies associées (cardiovasculaires, rénales, ORL, endocriniennes – thyroïde, diabète, maladie coeliaque – , etc.).
Le retard statural fait appel à l’hormone de croissance. Le traitement est initié par un médecin hospitalier habilité à sa prescription, lorsque la taille est ≤ -2 DS ou en cas de ralentissement statural important quel que soit l’âge (début en moyenne vers l’âge de 2 à 4 ans). la posologie recommandée est : 0,050mg/kg/jour, en une injection sous-cutanée quotidienne, le soir (0,35 mg/kg/semaine).
Concernant l’insuffisance hormonale on propose un traitement par estrogènes le plus souvent débuté à un âge pubertaire normal, vers l’âge de 12 ou 13 ans et/ou lorsque l’âge osseux est ≥ 11 ans , afin d’induire la puberté, hormis dans les rares cas de démarrage pubertaire spontané pouvant cependant nécessiter un traitement secondairement en cas de non progression pubertaire. La dose initiale correspond habituellement à environ 1/10ème de la dose de substitution œstrogénique de l’adulte (0,2 mg/jour de 17-bêta-œstradiol). Dans les formes diagnostiquées tardivement, certains préfèrent attendre 1 an de traitement par hormone de croissance pour débuter le traitement œstrogénique, afin d’optimiser la croissance prépubertaire sous GH. Le traitement est ensuite poursuivi par œstrogènes à faible dose au moins 2 ans afin de permettre un développement mammaire et utérin satisfaisants et d’éviter une progression excessive de la maturation osseuse. On augmente les doses d’œstrogènes à la fin de la période de croissance (vitesse de croissance < 2 cm/an) pour atteindre la dose de substitution adulte et introduction des progestatifs. L’adjonction d’un progestatif est cependant possible à partir d’une dose de 0,8 mg de 17-bêta-œstradiol/jour ou en cas de survenue de métrorragies sous œstrogènes à faibles doses. Ce traitement permettra la survenue des menstruations, qui surviennent en général dès la fin du 1er cycle sous traitement œstroprogestatif substitutif. A l’âge adulte le traitement est estroprogestatif soit avec de l’estradiol naturel, soit avec la pilule contracéptive.
Les grossesses spontanées sont exceptionnelles mais elles sont possibles par don d’ovocytes . Dans ce dernier cas, il y a une évaluation et préparation hormonale préalables de l’utérus . On réalise un bilan préalable si désir ou diagnostic de grossesse :
-cardio-vasculaire : PA, ECG, évaluation du calibre aortique (échographie, voire IRM) ;
-TSH, T4L, glycémie à jeun ;
– échographie pelvienne à réaliser avant la programmation de toute grossesse.
-Considérer toute grossesse comme à plus haut risque de complications et orienter la patiente vers une maternité de niveau 2 ou 3.
-Recherche de diabète pendant la grossesse.
-Surveillance cardio-vasculaire pendant toute la grossesse : mesure de la PA à chaque consultation, du calibre aortique par échographie à la fin des 1er et 2e trimestres et mensuellement lors du dernier trimestre, avec contrôle dans les 8 à 15 jours post-partum (réalisation d’une IRM aortique en fonction de l’avis cardiologique spécialisé). Informer d’un risque accru de fausse couche, de survenue d’anomalies chromosomiques en cas de grossesse spontanée avec possibilité de diagnostic prénatal.
b-Insuffisance ovarienne à caryotype normal 46XX. Dans ces cas, la taille est normale, les ovaires sont de petite taille. Certaines anomalies génétiques ont pu être identifiées : microdélétion du chromosome X, mutation du gène du récepteur de la FSH, mutation du gène Foxl2 (syndrome ptosis-blépharophymosis), prémutation du gène X Fra. Mais dans la majorité des cas, la cause n’est actuellement pas retrouvée.
c-Dysgénésies gonadiques pures 46XY. La dysgénésie gonadique à 46,XY est une anomalie du développement sexuel associée à un développement anormal des gonades qui se traduit par la présence d’organes génitaux féminins externes et internes malgré le caryotype masculin 46,XY.Le phénotype est féminin, les organes génitaux internes féminins, les gonades sont de petite taille mais indifférenciées et réduites à des bandelettes. Il y a un impubérisme avec aménorrhée malgré une pilosité axillopubienne normale. Ce type de gonades sont associées à un haut risque de tumeurs abdominales (dysgerminome le plus souvent) qui peuvent être inaugurales dans certains cas. Il n’y pas de signe de syndrome de Turner et la stature est normale ou au-delà.
L’étiologie n’est pas complètement élucidée; cependant la DG complète à 46,XY est due à un échec de la détermination testiculaire par interruption de son activation génétique en cascade et plusieurs gènes ont été impliqués: SRY (délétion ou mutations perte-de-fonction; Yp11.3), NR5A1 (9q33) et DHH (mutations homozygotes ou hétérozygotes composites; 12q13.1). De plus, chez des patientes ayant des duplications partielles du chromosome Xp (englobant le gène NR0B1) et des délétions du chromosome 9p (emportant les gènes DMRT1 et DMRT2), il existe aussi une DG complète à 46,XY isolée. Des mutations du gène CBX2 ont été rarement rapportées, en particulier chez une patiente avec développement de tissu ovarien malgré le caryotype masculin 46,XY. Des mutations du gène MAP3K1 (chromosome 5q), responsables d’anomalies en cascade de la voie de signalisation des MAP kinases, ont été récemment identifiées dans deux cas familiaux et deux cas sporadiques. Des facteurs environnementaux (prise de progestérone pendant la grossesse) et un retard de croissance prénatale ont aussi été associés à la DG complète à 46,XY.
Le diagnostic repose sur le tableau clinique, l’examen cytogénétique, le bilan hormonal, les analyses moléculaires et parfois l’exploration chirurgicale avec biopsie et gonadectomie.
Les diagnostics différentiels sont les dysgénésies ovariennes hypergonadotropes à 46,XX et toutes les formes syndromiques de dysgénésies gonadiques à 46,XY (par exemple: le syndrome de Frasier, la dysplasie campomélique et l’anomalie du développement sexuel à 46,XY avec insuffisance surrénalienne.
Le diagnostic prénatal est possible par l’analyse moléculaire si l’anomalie génétique a été identifiée dans la famille et il est recommandé dans les formes syndromiques. Bien que certains cas soient sporadiques, un conseil génétique peut être proposé aux familles et doit être adapté au mode de transmission de l’anomalie génétique identifiée.
La prise en charge nécessite une gonadectomie précoce à cause du risque élevé de transformation maligne et le traitement d’éventuelles anomalies associées selon la forme génétique. Une substitution hormonale est recommandée au moment de la puberté. Une aide psychologique doit aussi être proposée aux patientes et à leur famille. L’infertilité est un problème important dans la prise en charge; la grossesse est cependant possible par don d’ovocytes.
2-Insuffisances gonadiques acquises
a-Radiothérapie : l’insuffisance ovarienne est quasi constante si la dose d’irradiation ovarienne est de plus de 20 grays.
b-Chimiothérapie avec des agents alkylants et tout particulièrement après intensification comportant du busulphan ou des fortes doses de cyclophosphamide.
c-Auto-immune : le plus souvent associée à d’autres atteintes auto-immunes, en particulier dans le cadre de polyendocrinopathies auto-immunes multiples (candidose, hypothyroïdie, hypoparathyroïdie, insuffisance surrénalienne, diabète type 1).
d-Galactosémie : accumulation intracellulaire du galactose et de ses métabolites.
B-Insuffisances gonadotropes
Ce sont des cas où la cause est une insuffisance de stimulation des ovaires par les hormones hypophysaires FSH et LH.
1-Hypogonadisme hypogonadotrope permanent
a-Déficit gonadotrope isolé ou associé à des troubles de l’olfaction (anosmie ou hyposmie) dans le cadre d’un syndrome de Kallmann (anomalies de migration des cellules à LHRH et des cellules olfactives).
b-Syndromes malformatifs : syndrome de Prader-Willi (obésité, retard mental, retard statural, dysmorphie), syndrome de Laurence-Moon-Bardet-Biedl (obésité, retard mental, polydactylie, rétinite pigmentaire).
c-Insuffisance hypophysaire globale avec déficits somatotrope (GH) et parfois thyréotrope (TSH) ou corticotrope (ACTH) ; elle peut être :
-Tumorale : crâniopharyngiome (tumeur la plus fréquente de la région hypothalamo-hypophysaire responsable d’une hypertension intracrânienne, d’un retard statural), germinome, astrocytome et plus rarement adénome à prolactine ;
– Congénitale : syndrome d’interruption de la tige pituitaire avec le plus souvent un déficit en GH connu et traité depuis l’enfance ;
-Séquellaire d’une chirurgie ou d’une radiothérapie (irradiation hypophysaire > 30 Gy) pour tumeur de la région hypothalamo-hypophysaire.
2-Hypogonadisme hypogonadotrope fonctionnel transitoire
Toutes les pathologies chroniques avec répercussion nutritionnelle ou syndrome inflammatoire évoluant depuis l’enfance peuvent être la cause d’un retard pubertaire. Mais le retard pubertaire peut parfois révéler la maladie chronique ; c’est le cas en particulier de la maladie de Crohn ou la maladie cœliaque. C’est aussi le cas de l’anorexie mentale.
C-Le retard pubertaire simple
Bien sûr, il existe des retards pubertaires simples qui vont se résoudre naturellement. Le retard pubertaire simple reste toutefois un diagnostic d’élimination. Il existe souvent un ralentissement statural associé, un retard d’âge osseux et des antécédents de puberté tardive dans la famille. Il s’agit de cas extrêmes du développement pubertaire normal. Ce type de retard correspond aux 2,5 % des sujets qui déclencheront leur puberté après 14 ans, chez le garçon, ou après 13 ans, chez la fille. Le retard pubertaire simple pose un problème diagnostique difficile avec certains hypogonadismes hypogonadotrophiques partiels ou isolés.