I-L’ascite et l’anasarque foeto-placentaire
A-Description
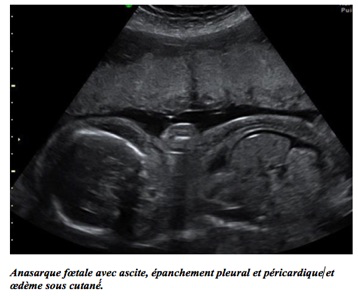
L’ascite minime se traduit par un simple croissant liquidien séparant le contour hépatique de la paroi abdominale antérieure. En cas d’ascite importante, le foie flotte autour de la portion péritonéale de la veine ombilicale et les anses digestives sont refoulées en arrière autour du rachis.
L’anasarque foetoplacentaire associe :
– une ascite importante avec hydrocèle de la vaginale du testicule et souvent une hépatomégalie ;
– un hydrothorax parfois associé à un épanchement péricardique ;
– un œdème sous-cutané plus ou moins diffus avec aspect de double contour entourant le tronc et pôle céphalique, ou un hygroma kystique au premier trimestre (syndrome de Bonnevie-Ullrich) ;
– un hydramnios et un œdème du cordon ;
– un épaississement du placenta, variable selon les étiologies.
B-Causes
Les causes d’une simple ascite sont le plus souvent : maladie hépatique, perforation intestinale, maladie pancréatique, Cardiopathie,maladies métaboliques par déficit enzymatique. On considère cependant que l’ascite isolée et l’anasarque répondent aux mêmes étiologies.
1-Les causes cardiaques :
-Malformations l’hypoplasie ventriculaire gauche ou droite, le canal atrio-ventriculaire…
-Arythmie (qui n’est pas toujours causée par des lésions anatomiques identifiables) comme la tachyarythmie et la bradyarythmie qui peut être provoquée par un bloc auriculo-ventriculaire complet ou une maladie maternelle (lupus, polyarthrite).
-Insuffisance cardiaque avec débit cardiaque élevé qui peut être dû à un chorioangiome placentaire ou un tératome sacrococcygien.
-Cardiomyopathies et rhabdomyome cardiaque.
2-Les causes chromosomiques
Syndrome de Turner – 45X, trisomies 21 et 18,riploïdie , trisomie 13, 15, 16, tétraploïdie.
3-L’anémie fœtale
qui peut se confirmer rapidement par l’étude vélocimétrique au niveau de l’artère cérébrale moyenne et dont les causes sont :
-Isoimmunisation
-Alpha-thalassémie homozygote (55 % des cas)
-Maladies hématologiques (hémoglobinopathies, dysérythropoièse…)
-Enzymopathie
-G-6PD
-Parvovirus B 19
Transfusion foeto-maternelle
-Hémorragie intra-fœtale
4-Les infections fœtales
pour lesquelles l’anasarque peut être une anomalie transitoire :
-Cytomegalovirus
-syphilis (ascite).
5-Les causes pulmonaires et thoraciques :
-Maladie kystique du poumon
-Séquestration pulmonaire
-Hernie diaphragmatique droite
-Tératome / Tumeur intra-thoracique
-Kystes bronchogéniques
-Chondrodysplasie
-Atrésie laryngée avec distension pulmonaire congénitale
-Chylothorax / Hydrothorax
6-Syndrome transfuseur-transfusé:
Le fœtus transfusé présente une hypervolémie avec insuffisance cardiaque responsable des épanchements.
7-les causes plus rares
-Toxoplasmose, rubéole, herpès, listeria, diabète (rare++)
-Malformation des voies urogénitales / pathologie intestinale ou hépatique :
-Akinésie fœtale / hypomobilité.
-Maladies lysosomales : représentant 1/5000 naissances, elles sont une cause non négligeable d’anasarque. La liste ci-dessous n’est pas exhaustive :
-Maladie de Niemann-Pick
-Maladie de Gaucher type 2
-Syndrome de Smith-Lemli-Opitz (défaut dans le métabolisme du cholestérol)
-Maladie de Farber
-Maladie de Wolman
-Syndrome de Hurler
-GM1 Gangliosidose Type I
-Hémochromatose
-Mucopolysaccharidose IV, VI et VII (déficit en beta-glucuronidase)
-Galactosialidose (déficit en neuraminidase et beta-galactosidase)
-Sialidose infantile ou mucolipidose type I & II (déficit en neuraminidase)
-Glycogénose II (maladie de Pompe), IIb (maladie de Danon), IV (maladie d’Andersen)
8-les causes du cas particulier de l’hygroma kystique
– Causes chromosomiques (Turner)
-Causes non chromosomiques comme les syndromes de Noonan, Fryns, Brachmann-de-Lange, Fraser, syndrome des pterygium multiples, l’achondrogénèse…
C-Conduite à tenir
-Échographie complète et détaillée du fœtus et du placenta
-Échocardiographie fœtale
-Doppler de l’artère cérébral moyenne
En fonction des résultats de ces examens il y a lieu de décider d’un éventuel traitement en urgence. Si ce n’est pas le cas, une amniocentèse est réalisée pour caryotype et mise en culture.
Du liquide doit être conserér pour d’éventuelles analyses complémentaires : analyses métaboliques (Gaucher, Tay-Sachs, GM1 Gangliosidose), analyse du surnageant (mucopolysaccharides, oligosaccharides, acide sialique libre et mesure de l’activité hydrolasique acide), études moléculaires (désordres mitochondriaux et métaboliques).
Eventuellement, analyse de l’épanchement fœtal (liquide ascitique et pleural) pour connaître le taux de protéine, le contenu ADN de la cellule et la numération des lymphocytes
L’examen anatomo-pathologique du fœtus et du placenta est indispensable pour le diagnostic étiologique et le conseil génétique.
II-Lorsque l’estomac est non visible
Un estomac non visible, sur plusieurs examens successifs, doit évoquer une atrésie de l’œsophage ou une hernie diaphragmatique gauche.
A-L’atrésie de l’œsophage (1 pour 5000 naissances) associe :
– hydramnios présent dans 85 % des cas et important. C’est souvent le signe d’appel des formes sans fistule trachéo-œsophagienne. Dans les formes avec fistule l’hydramnios est moins fréquent (32 %) et moins important ;
– non-visibilité de l’estomac à deux examens successifs, traduisant le non-remplissage. Cependant, l’estomac vide peut être parfois deviné du fait d’un léger remplissage peut se produire par la fistule œso-trachéale ou par les sécrétions de la paroi gastrique.
Il faut rechercher des anomalies associées car elles sont fréquentes (40 à 60 % des cas) et elles alourdissent le pronostic. Les plus fréquentes sont cardiaques, ano-rectales, génito- urinaires et vertébrales. Elles entrent souvent dans le cadre de syndromes dont le VACTERL (Vertebral defects, Ano-rectal anomalies, Cardiac malformations, Tracheo Esophageal fistula, Renal anomaly et Limb pour dysplasie radiale).
Les anomalies chromosomiques ne sont pas non plus rares (environ 5 à 10 % des cas : trisomie 18 et trisomie 21), le caryotype doit donc être systématique. La plupart des atrésies sont cependant sporadiques et le risque de récurrence est faible.
B-Hernie diaphragmatique gauche
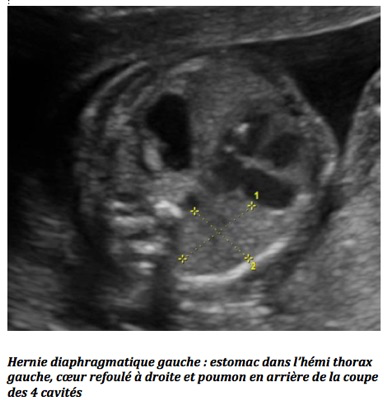
Elle est quatre à cinq fois plus fréquente qu’à droite et concerne 1/2 200 naissances. À l’étage abdominal, l’estomac n’est pas visible, la veine ombilicale est déviée, le périmètre abdominal est petit. À l’étage thoracique, on individualise une image liquidienne en arrière du cœur qui peut être dévié à droite et dont les battements lui sont transmis. Après 30 SA, les clartés du tube digestif hernié peuvent être visibles en intra-thoracique.
Il faut rechercher des malformations associées : cœur, système nerveux central, paroi abdominale (omphalocèle), système urinaire. Le syndrome de Fryns, par exemple, comporte une hernie diaphragmatique, des anomalies de la face et des extrémités.
On retrouve parfois des anomalies chromosomiques associées concernant surtout la trisomie 18.
Une position centrale ou inhabituelle de l’estomac dans l’abdomen doit faire rechercher une hernie diaphragmatique droite, une éventration diaphragmatique ou une hernie hiatale. Un estomac situé plus volontiers sur la droite peut évoquer un situs abdominal inversé plus ou moins complet qui, associé à une anomalie cardiaque, est retrouvé dans un syndrome cardio-splénique tel le syndrome d’Ivemark (avec asplénie caractéristique de la forme complète ou polysplénie).
III-Si la vessie n’est pas visible
L’absence de vessie sur plusieurs examens successifs, évoque :
– en présence d’un oligoamnios sévère, un défaut de production urinaire par agénésie rénale bilatérale ou obstruction bilatérale haute et l’étude des loges rénales précise le diagnostic ;
– en présence de liquide amniotique normal ou augmenté, une malformation vésicale qui se résume à l’exstrophie vésicale
en présence de liquide amniotique normal ou augmenté, une malformation vésicale qui se résume à l’exstrophie vésicale.
IV- Vascularisation ombilico-portale anormale
Des modifications de cet axe vasculaire sont assez fréquemment retrouvées et peuvent traduire :
1-Une persistance de la veine ombilicale droite
L’axe ombilico-portal s’incurve vers la gauche (et non vers la droite) et la vésicule biliaire se situe à gauche de celui-ci. Sans conséquence quand elle est isolée, elle peut être néanmoins retrouvée dans de nombreuses anomalies morphologiques notamment cardio-vasculaires et digestives.
2-Une agénésie du canal d’Arantius
On peut rencontrer deux situations:
– soit le flux ombilical court-circuite complètement le foie. La veine ombilicale gauche ne s’anastomose pas avec le système veineux omphalo-mésentérique mais s’abouche directement dans l’oreillette droite, la veine cave inférieure, une veine rénale ou iliaque. Le canal d’Arantius est inconstamment présent. La perfusion et l’oxygénation hépatique sont réduites et le cœur hyperperfusé. Cette situation qui est la plus fréquente est de diagnostic facile
– soit le flux ombilical court-circuite complètement le foie. La veine ombilicale gauche ne s’anastomose pas avec le système veineux omphalo-mésentérique mais s’abouche directement dans l’oreillette droite, la veine cave inférieure, une veine rénale ou iliaque. Le canal d’Arantius est inconstamment présent. La perfusion et l’oxygénation hépatique sont réduites et le cœur hyperperfusé. Cette situation qui est la plus fréquente est de diagnostic facile
3- Une ectasie de la veine ombilicale
– isolée, les conséquences anténatales sont nulles mais un contrôle cardiaque postnatal fonctionnel est nécessaire ;
– associée à d’autres malformations, il faut penser à rechercher une anomalie chromosomique (10 %).
4- Une fistule vasculaire intra-hépatique:
Son diagnostic est facilité par le Doppler couleur qui individualise le shunt vasculaire où il existe un flux turbulent avec aliasing. Souvent isolée, il faut néanmoins vérifier qu’elle ne soit pas le témoin d’une tumeur hépatique. La surveillance est poursuivie en anté- puis en postnatal à la recherche d’une décompensation cardio-vasculaire. La fermeture du shunt vasculaire peut être spontanée ou nécessiter une embolisation.
5- Un axe trop oblique vers le haut :
Il faut penser à vérifier l’absence de hernie diaphragmatique droite.
V-Les images liquidiennes anormales
A-Dilatation d’un repère liquidien
1-Dilatation de l’estomac
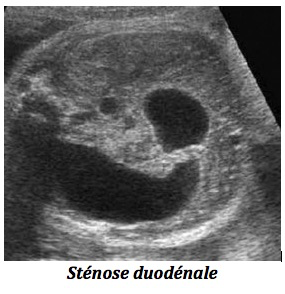
-Surtout la sténose duodénale (1/5000) naissances ; elle est stable à deux examens successifs et un hydramnios est associé. Le bilan doit comporter une échographie détaillée à la recherche de malformations associées fréquentes notamment de cardiopathies, d’autres anomalies digestives (atrésie de l’œsophage), d’anomalies squelettiques, vertébrales ou génito-urinaires. La réalisation d’un caryotype sera systématique pour rechercher, avant tout, une trisomie 21 retrouvée dans un tiers des cas. Le pronostic est bon quand l’anomalie est isolée. La prématurité est fréquente, conséquence de l’hydramnios. La prise en charge pédiatrique et chirurgicale précoce est indispensable.
-Une dilatation de l’estomac anormale peut également évoquer l’exceptionnelle atrésie du pylore qui, associée à une épidermolyse bulleuse, réalise le syndrome de Carmi (génodermatose autosomique récessive, létale dans la 1re année de vie.
2-Dilatation vésicale excessive
Reconnue à sa position médiane, la visualisation en Doppler couleur des artères ombilicales de part et d’autre de cette image liquidienne, et une dilatation pyélique et urétérale plus ou moins rapidement associée, ou parfois une dysplasie rénale bilatérale. Elle répond à trois causes principales :
– la mégavessie de lutte, en amont d’un obstacle urétral formé le plus souvent par les valves de l’urètre postérieur, surtout chez le garçon, caractérisé par la dilatation de l’urètre réalisant le récessus urétral
– le syndrome de prune belly avec absence de musculature abdominale, grande vessie hypotonique, parfois hypotonie pariétale visible réalisant une pseudo-éventration, cryptorchidie chez le garçon
– le syndrome mégavessie-microcolon-hypopéristaltisme intestinal qui sera évoqué chez une fille (90 % des cas) présentant une vessie volumineuse avec hydronéphrose bilatérale et estomac bien vu. La quantité de liquide amniotique est normale ou même augmentée. Le pronostic est très mauvais avec seulement 11 % de survivants à long terme.
B-Images liquidiennes surajoutées
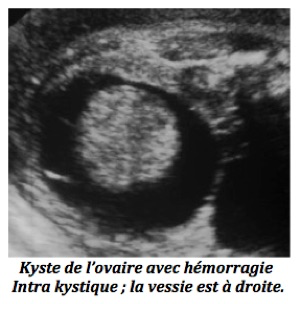
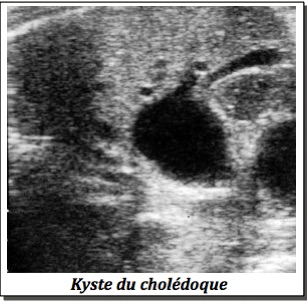
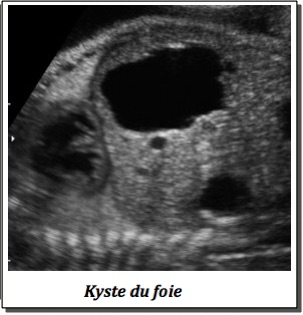
1-En cas d’image kystique solitaire,
on évoque en fonction de la localisation :
a-En position médiane
-Kyste de l’ovaire. il est généralement purement liquidien mais parfois échogène ce qui fait évoquer un saignement intrakystique ou une torsion. Il s’agit souvent, mais pas toujours, d’un kyste fonctionnel qui régressera après la naissance.
-Kyste de l’ouraque
b-Dans l’hypochondre droit
-Kyste du cholédoque.
-Kyste hépatique.
c-Dans l’hypochondre gauche
-Kyste splénique
-Lymphangiome
-Kyste épidermoïde
d-Dans les flancs ou les fosses lombaires
-Kyste du rein ou de la surrénale
-Kyste du mésentère
-Duplication digestive
-Diverticule de Meckel géant
2-En cas d’images liquidiennes multiples ou tubuliformes
a-Sténoses et obstructions digestives
-Les obstructions de l’intestin grêle, sont les plus fréquentes et elles sont faciles à voir, donnant des images liquidiennes multiples, irrégulières et plutôt centrales. L’existence d’un péristaltisme est bien sûr évocatrice ; l’hydramnios est inconstant. L’évolution de l’obstruction sera évaluée par des échographies itératives : l’augmentation du diamètre des anses, la raréfaction du péristaltisme, l’apparition de cloisons, niveaux liquides et débris traduisant un remaniement nécrotico-hémorragique de l’intestin, l’apparition d’une ascite ou de signes de péritonite méconiale constituent des critères de dégradation.
-À signaler, l’apple peel syndrome, pathologie autosomique récessive qui correspond à une atrésie du grêle par occlusion de l’artère mésentérique supérieure ou de l’une de ses branches.
-Les occlusions coliques et ano-rectales sont plus rares et de diagnostic beaucoup plus difficile. La muqueuse colique reste en effet active et résorbe les sécrétions intestinales. L’aspect du côlon est le plus souvent normal, tout au plus peut-on noter une « trop belle image colique », persistante au contrôle. Ce signe serait assez particulier à la maladie de Hirschprung, du fait des troubles de réabsorption du liquide colique.
La possibilité de la persistance d’un cloaque est évoquée devant la complexité des images pelvi-abdominales avec mise en évidence d’une structure liquidienne anormale dont le volume peut se modifier en fonction de la vidange vésicale.
b-Rein multikystique
On ne retrouve pas de parenchyme rénal normal et les kystes multiples ne communiquent pas entre eux, ce qui permet, en théorie, de faire la différence avec une hydronéphrose évoluée
c-Lymphangiome kystique du mésentère
L’iimage est multiloculaire, de taille variable, à paroi fine, non vascularisée, refoulant les anses intestinales. Certains kystes peuvent saigner entraînant la formation de caillots se traduisant par des zones échogènes diffuses au sein du lymphangiome.
VI-Les images hyperéchogènes
A-Intestin hyperéchogène
L’appréciation de l’hyperéchogénicité intestinale globale (paroi et contenu) est assez subjective et nécessite des points de repère. Nyberg et Slotnick proposent une classification pratique :
– grade 0 : intestin isoéchogène au foie (normal) ;
– grade 1 : échogénicité plus marquée mais inférieure à celle de l’os iliaque
– grade 2 : échogénicité identique à celle de l’os iliaque
– grade 3 : échogénicité supérieure à celle de l’os iliaque.
La découverte d’un grêle hyperéchogène (grade 2 et 3), stable et persistant après 20-22 SA nécessite :
– un bilan morphologique complet pour apprécier son caractère isolé ou non ;
– une étude biométrique et vélocimétrique pour rechercher un retard de croissance intra-utérin ;
– la réalisation d’un caryotype ;
– la recherche d’une fœtopathie infectieuse, en particulier cytomégalovirus et toxoplasmose
– la recherche d’une mucoviscidose
B-Iléus méconial
il se caractérise par un intestin grêle agglutiné sous forme d’une petite masse abdominale hyperéchogène siégeant souvent en fosse iliaque droite et associée à une dilatation des anses grêles en amont. En cas d’iléus méconial après 20 SA, une mucoviscidose doit être recherchée systématiquement, même en l’absence de tout antécédent familial. Le bilan comprend une amniocentèse avec dosage des enzymes digestives (gamma-GT, LAP, phosphatases alcalines), ainsi qu’une ponction de sang fœtal, avec étude de l’ADN fœtal pour rechercher les mutations les plus courantes (delta F 508 sur le chromosome 7), parmi les onze mutations connues. En cas d’antécédents familiaux, le diagnostic de mucoviscidose peut être réalisé précocement par prélèvement de villosités choriales dès 12 semaines ou par amniocentèse précoce avec étude enzymatique.
C-Péritonite méconiale
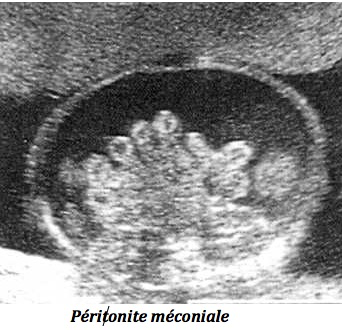
Elle est secondaire à une perforation digestive : le méconium et les enzymes digestives (trypsine) entraînent une réaction inflammatoire intense aboutissant à une péritonite aseptique. Les signes sont progressifs dans le temps : ascite échogène, puis avec échos denses le long de la paroi abdominale, puis enkystement sous forme de pseudo-kystes calcifiés.
D-Zones échogènes intra-hépatiques
Fréquentes (1/1 500 grossesses), elles sont le plus souvent banales et sans conséquence clinique : le bilan étiologique est alors négatif. Cependant elles traduisent parfois un processus infectieux qui sera recherché systématiquement. Elles s’associent plus rarement à un processus tumoral.
E-Vésicule biliaire hyperéchogène
Une vésicule biliaire de taille normale peut contenir des amas échogènes diffus ou en motte, ou parfois être complètement remplie d’échos, voire présenter un aspect lithiasique. Cet aspect de cause inconnue, est habituellement sans conséquence ultérieure et disparaît dans les premières semaines de vie.
F-Gros reins hyperéchogènes
La polykystose rénale de type infantile, autosomique récessive, en est l’étiologie principale. Elle donne un aspect de gros reins, très échogènes, du fait des multiples interfaces liées aux microkystes. La sémiologie échographique est assez caractéristique par le caractère bilatéral, la grosse taille des reins et l’oligoamnios, d’apparition plus ou moins précoce, traduisant une altération de la fonction rénale.
G-Tumeurs abdominales
-Néphroblastome
-neuroblastome surrénalien
-Tumeurs neurogènes postérieures
-Séquestrations pulmonaires (proches du diaphragme)
-Extension abdominale d’un tératome sacrococcygien.